
Skeletal Muscle Calcium Channel Mutation R528G: Enhanced Channel Inactivation and Omega-Current at Hyperpolarization Contribute to Hypokalemic Periodic Paralysis.
Abstract
Autosomal dominant inherited hypokalemic periodic paralysis (HypoPP) is caused by S4 voltage sensor mutations in skeletal muscle CaV1.1 calcium or NaV1.4 sodium channels. In the present study, a small German family with the known CaV1.1-R528G is described. The phenotype consists of short and infrequent episodes of limb weakness with ictal respiratory and cardiac involvement. There is incomplete penetrance in women, and acetazolamide is beneficial in two patients also taking daily potassium. Expression of the mutation in the GLT mouse muscle cell line revealed accelerated kinetics of inactivation by twofold, a left-shift of the steady-state inactivation curve by 13mV and a reduced recovery from fast inactivation by up to 39%. These changes suggest a stabilization of the inactivated state. Additional significant slowing of activation may support a second open state with differing ion selectivity or decreased activation of calcium-activated potassium channels and thereby contribute to weakness similar to other CaV1.1 mutations. Also, as documented for other HypoPP mutants, we found a hyperpolarization-induced inward guanidinium current of 22nS/nF which can be interpreted as an omega current along the voltage sensor gating pore that leads to a gain- of- function at potentials near the resting membrane potential. This finding can explain the long-lasting depolarizations that are known to lead to paralysis. The omega current is large enough so that a relatively mild hypokalemic trigger of 2.4mM already produces episodes of weakness in vivo.
Article Information
- Received
- Accepted
- Published
Academic Editor: Zheng Jiang, Johns Hopkins University School of Medicine
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2016 Marcin Bednarz, et al.
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Corresponding author: Karin Jurkat-Rott, Email:, Division of Neurophysiology, Ulm University, Albert-Einstein-Allee, 11, 89081 Ulm —
Competing Interests
The authors have declared that no competing interests exist.
Funding
No specific funding statement was provided by the authors.
Data Availability
No data-availability statement was provided by the authors.
Acknowledgements
This study was supported by the Else-Kröner Fresenius Foundation, the IonNeurONet of the German Federal Ministry of Research BMBF, the German Muscle Disease Society DGB and the non-profit Hertie Foundation.
Citation:
Introduction
Familial hypokalemic periodic paralysis (HypoPP) is an autosomal dominant disorder of the skeletal muscle. It is characterized by episodes of generalized paralysis caused by reduced serum potassium. Additional permanent weakness and myopathy occurs in 68% of patients 1. Two genes of voltage-gated cation channels are genetically causative, CACNA1S coding for CaV1.1 (HypoPP-1) and SCN4A encoding NaV1.4 (HypoPP-2). The CaV1.1 Ca2+ channel serves both as ion-conducting pore and as voltage sensor for excitation-contraction coupling while NaV1.4 initiates the action potential. Almost all mutations are located in the voltage-sensing transmembrane segment S4 and replace arginine residues occurring at every third position (reviewed in 2, 3, 4). Glycine mutations generally reduce treatment response to the standard medication acetazolamide 5.
Pathogenetically, a long-lasting membrane depolarization is responsible for the paralysis episodes and has been shown to be caused by an aberrant omega current that flows through the S4 mutation gating pore instead of the central ion-conducting alpha pore of the channel (originally described in 6, 7). Depending on the location of the mutated residue, the voltage range in which the omega current occurs differs because S4 segments move outward during channel activation. Briefly, the more inward the mutation the more depolarized the voltage range; therefore, the outermost mutations conduct omega currents at hyperpolarized potentials when the central alpha pore is closed (reviewed in 2, 3, 4). Also, the size of the residue replacing arginine influences the size of the omega current suggesting arginine-to-glycine mutations to generate the largest omega currents and therefore the most severe phenotypes (reviewed in 8).
Even though almost all HypoPP functional studies have been done on NaV1.4 mutations, CaV1.1 mutations are much more abundant and explain up to 77% of patients with hereditary HypoPP 9 with the most frequent being R528H and R1239H 10. It has not been systematically shown that the conclusions drawn for NaV1.4 are applicable to CaV1.1. Until now, there is only circumstantial evidence for omega currents of R1239H in native muscle of patients 11 and measurements of omega current of R528 in native muscle of transgenic mice 12. Additionally, our group detected omega currents using a mouse cell line expressing R1242G-CaV1.1 which is, however, associated to normokalemic periodic paralysis with transient compartment-like syndrome 13. For several other mutations in CaV1.1, omega currents and alpha pore currents have not yet been studied.
In this work, we study the R528G mutation in domain II of CaV1.1 for which neither omega current nor alpha pore current has been examined. It has been reported in a one large HypoPP family 14 and as a de novo mutation in a single patient 15. In this study we found it in a small German family. Based on the current model of pathogenesis, we would expect i) a more severe phenotype, ii) a bad response to acetazolamide and iii) a large omega current in the range of the resting membrane potential as pathogenetic mechanism. We test these hypotheses and additionally examine alpha pore currents to discuss their possible contribution to the phenotype.
Patients and Methods
Patients
Clinical and genetic examination was approved of by the institutional review board of Ulm and conducted according to the declaration of Helsinki. Blood was taken with informed consent from the proband, his brother, mother, and aunt. His mother sent blood but did not wish to be examined otherwise. Exons 11 and 30 of CACNA1S were studied for routine diagnosis using Sanger sequencing.
Mutagenesis
Rabbit cDNA in a PSG5 vector (Stratagene) containing pGFP37 was employed for site-directed mutagenesis 13. The C1582G cDNA mutation which results in the R528G amino acid change was introduced using PCR-based site-directed mutagenesis with the reverse oligonucleotide 5’-CGGATGCAGTGCAACACGG-3’ (Eurofins MWG GmbH, Ebersberg) and verified by Sanger sequencing. Heterologously expressed rabbit cDNA of CaV1.1 in GLT cells produces larger currents with otherwise equivalent gating parameters to human cDNA as shown previously 16.
Cell Culture and Transfection
Myotubes of the homozygous dysgenic cell line GLT were cultured as previously described 13, 16, 17. Briefly, GLT cells were cultured in a growth medium consisting of 80% Dulbecco's modified Eagle medium, 10% fetal bovine serum, and 10% horse serum (HS), 1% Glutamate (all Gibco) in sterile flasks or gelatine-coated dishes. Serum content was reduced to fusion medium containing only 2% HS, on day 1 after the last passaging procedure to induce maturation and fusion of cells in the dishes. Transfection was performed with 4µg FugeneHD (Roche) reagent to 2µg cDNA on cells, two days after first use of fusion medium.
Electrophysiology
Standard whole-cell recordings were performed on 7-9 days old myotubes. Series resistance was partially (30-60%) compensated by the analog circuitry of the Axopatch 200B patch-clamp amplifier (Axon Instruments), with voltage errors less than 5 mV. Before acquiring data, individual cells were allowed to equilibrate for 5min after achieving internal access. For alpha pore current experiments, the bathing solution contained (in mM): TEA-Cl 132, CaCl2 10, MgCl2 1, HEPES 10, glucose 5, 4-aminopyridine (4-AP) 2.5, tetrodotoxin 0.005 (pH 7.4). Pipettes were filled with (in mM): CsCl 128, HEPES 12, EGTA 10, Mg-ATP 5, phosphocreatine 5 (pH 7.2). For omega current measurements, the bathing solution contained (in mM): TEA-methanesulfonate (MS) 80, Guanidinium sulfate 38, MgSO4 1, Ca-gluconate 5, Glucose 5, 4-aminopyridine (4-AP) 2.5, HEPES 10 (pH 7.4). Pipettes were filled with (in mM): TEA-MS 65, Cs-MS 65, EGTA 10, MgATP 5, Na2CreatinPO4 5, HEPES 12 (pH 7.2). All measurements were performed at room temperature (20-25℃). Data were filtered at 1kHz and sampled at 2kHz. Each experiment was performed on six to ten cells.
Pulse Protocols for Alpha Currents
To elicit whole-cell Ca2+ currents, a series of 600ms pulses from -60mV to 50mV in 5mV steps from a holding potential of -90mV were performed. Steady-state activation parameters were determined by fitting the current-voltage relation with the equation I=Gleak*(V-Vleak)+Gmax*(V-Vrev)/(1+exp((V0.5-V)/k)), where Gleak and Vleak respectively are the conductance and reversial potential of the linear leak, Gmax and Vrev respectively are maximum conductance and reversal potential of the alpha pore current V0.5 and V respectively are the potential for the half-maximal current and the test potential, and k is the slope factor. The kinetics of activation was estimated by fitting the activation time course to a single exponential function as I=exp(−t/τact)+C and plotting the time constant of activation (τact) as a function of the test voltage ranging between +10mV and +50mV. To avoid interference by a residual capacitive transient, we began the fits 8ms after the start of the test pulse.
Figure 1. CaV1.1 alpha pore currents in GLT myotubes. Original traces of calcium alpha pore currents from heterologously expressed wild-type (A) and R528G (B) channels in GLT myotubes. (C) Conductance-voltage relationship from Bolzmann fits of steady-state activation and inactivation. Note the left shift of inactivation of the mutant. (D) Kinetcs of activation and (E) inactivation determined by monoexponentional fits. Note the slowed activation and accelerated inactivation of the mutant. (F) Recovery from fast inactivation as determined by a single exponential fit. Note the incomplete recovery of the mutant. All error bars represent the mean ± SEM. The p values are *<0.05; **<0.01 and ***<0.001.
Download figure
For the examination of the steady-state activation, cells were held at -90mV and then depolarized by a series of 60s prepulses from -90mV to +30mV in 10mV increments prior to a test pulse of 30mV. The peak Ca2+ current measured during the test pulse was plotted as a function of conditioning voltage for both WT and R528G channels (Figure 1C). Boltzmann functions were fitted to the data of the WT and mutant channels by the equation I/Imax=A/(1+exp((V0.5-V)/k))+C, where A represents the fraction of inactivated channels, V is the pre-pulse potential, V0.5 is the potential at which half of the channels are inactivated, C is the fraction of non-activated channels and k is the slope factor. The time constant of inactivation (τh) was determined by fitting a single exponential function to the decaying part of the calcium currents as I=exp(−t/τh)+C and τh values were also plotted as a function of voltage.
The rate of recovery from inactivation at hyperpolarized voltages (-90mV, -80mV or -70mV) was measured by employing a double pulse protocol with varying intermediate repolarization times. Generally, an inactivating 20s prepulse to 30mV was followed by a 300ms test pulse to the same potential. The peak Ica was normalized to the peak current amplitude measured during the prepulse. Test currents were recorded in both WT and R528G cells after recovery intervals ranging from 0.2ms to 247ms at -90mV, -80mV and -70mV. The recovery from fast inactivation was analyzed by fitting the data to a single exponential function, I/Imax=A*(1−exp(−t/τ))+C, where A is the fractional amplitude, τ is the time constant, and C is the level of non-inactivating sodium current.
Pulse protocol for Omega Currents
The voltage dependence of omega currents was determined using 10mV steps from -140mV to 0mV over a period of 200ms from a holding potential of -90mV using solutions containing guanidinium ions. The linear leak was subtracted offline and calculated from a linear fitting curve between -40mV and 0mV using a linear fitting curve y=m*x+b between -40mV and 0mV. After leak subtraction, the current densities (mean current amplitudes divided by cell capacitance) were plotted against the eliciting voltage step. Omega currents were then determined by subtraction of currents from untransfected cells (unspecific background currents) from both WT and mutant currents for comparison.
Statistics
Whole-cell recordings were analyzed by a combination of pClamp (Axon Instruments), Excel (Microsoft) and ORIGIN (Microcal software, Northampton, MA) programs. Data are presented as mean± SEM, which is shown as bars. Student’s double-sided t-test was applied for equal variances or Welch t-test for unequal variances and unequal sample sizes. The significance level was set with *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001.
Results
Patients
All 4 samples of the family members we investigated, contained C1582G in CACNA1S, which codes for R528G in CaV1.1. The proband was referred because of tetraplegia with ictal bradycardia and a serum potassium level of 2.1mM at age 16. In addition to chronic recurrent myalgia, cramping and muscular weakness in the morning hours, the proband suffered from episodic incomplete paresis in the proximal muscle groups of the upper and lower extremities which decreased within hours. To treat a mild muscle weakness after exercise, he prophylactically takes oral potassium. No serious paralysis episodes have occurred after administering acetazolamide 250mg daily. Chronic muscle weakness has not present up to his current age of 26.
The proband’s younger brother reported that since approximately 3/4 of a year (onset at age 16), very occasionally a light paresis in the upper arm appears which he noted after prolonged exercise, especially in the evening hours. He takes no acetazolamide, but just lays down for sleep and the next morning it´s all gone. There is no chronic muscle weakness or any further symptoms especially in any facial or pharyngeal muscles.
The proband’s aunt had her first episode at age four and reported ictal cardiac arrhythmia. Her episodes of limb paralysis were rare, i.e. detected more or less yearly and lasted for minutes to hours. She also suffered from frequent painful muscle cramping. Her treatment was a combination of daily potassium supplementation plus 250mg acetazolamide. In contrast, the proband’s mother (sister of the aunt) reported herself to be unaffected, i.e. not having episodic or chronic weakness. She did not agree to a physical examination or provide any further information.
Functional Expression Levels
Successfully transfected cells were indicated by GFP fluorescence marking. Since measuring fluorescent density of the very variably shaped, multi-nuclear GLT myotubes was not reliable, maximal conductance, Gmax (as defined by the formula in the methods section) was used as an indicator of the amount of expressed channels instead. This is a common practice since the GLT cell line does not express endogenous, functional CaV1.1 17 and therefore, all alpha pore currents are due to plasmid expression. The Gmax values as indicated in Table 1 showed no significant difference between WT and mutant. Therefore, we considered their expression levels to be similar.
Table 1. Parameters of calcium alpha pore currents| WT | R528G | p | |
| Current activation | |||
| Gmax (pA/mV) | 23.34 ± 2.23 | 29.85± 8.53 | 0.48 |
| Vrev (mV) | 84.06 ± 4.24 | 78.93 ± 4.63 | 0.42 |
| V0.5 (mV) | 20.25 ± 1.94 | 16.51 ± 2.52 | 0.26 |
| k (mV) | 7.51 ± 0.34 | 7.46 ± 0.58 | 0.94 |
| Maximum current density (pA/pF) | 5.04 ± 1.17 | 7 ± 1.94 | 0.41 |
| n | 15 | 8 | |
| Activation time-constant (τa) | |||
| 0 (mV) | 10.28 ± 1.23 | 13.14 ± 1.5 | 0.16 |
| 10 (mV) | 14.66 ± 1.72 | 19.32 ± 1.92 | 0.1 |
| 20 (mV) | 16.68 ± 1.9 | 24.68 ± 2.81* | 0.03 |
| 30 (mV) | 15.71 ± 1.46 | 27.15 ± 4.22* | 0.02 |
| 40 (mV) | 13.93 ± 1.38 | 26.06 ± 4.25* | 0.01 |
| 50 (mV) | 12.2 ± 1.47 | 22.94 ± 4.02* | 0.02 |
| n | 7 | 6 | |
| Inactivation time-constant (τh) | |||
| 10 (mV) | 2.75 ± 0.37 | 1.25 ± 0.16** | 0.0012 |
| 20 (mV) | 1.81 ± 0.27 | 0.70 ± 0.07** | 0.0019 |
| 30 (mV) | 1.59 ± 0.14 | 0.63 ± 0.04**** | 0.000002 |
| 40 (mV) | 1.37 ± 0.15 | 0.61 ± 0.04*** | 0.00076 |
| n | 7 | 6 | |
| Steady-state inactivation | |||
| A | 0.97 ± 0.01 | 0.99 ± 0.01 | 0.12 |
| V0.5 (mV) | -23.33 ± 3.66 | -36.98 ± 4.71* | 0.045 |
| k (mV) | -11.45 ± 1.27 | -12.59 ± 0.71 | 0.45 |
| c | 0.01 ± 0.01 | 0.01 ± 0.01 | 0.95 |
| n | 6 | 6 | |
| Recovery from inactivation | |||
| -90mV | |||
| A | 0.79 ± 0.03 | 0.78 ± 0.04 | 0.8 |
 1 (ms) 1 (ms) |
26.05 ± 2.93 | 39.44 ± 6.21 | 0.06 |
| c | 0.06 ± 0.02 | 0.05 ± 0.03 | 0.91 |
| n | 7 | 6 | |
| -80 mV | |||
| A | 0.74 ± 0.02 | 0.61 ± 0.02** | 0.003 |
 1 (ms) 1 (ms) |
29.69 ± 3.67 | 40.72 ± 6.52 | 0.17 |
| c | 0.01 ± 0.01 | 0.03 ± 0.02 | 0.33 |
| n | 6 | 6 | |
| -70mV | |||
| A | 0.69 ± 0.02 | 0.42 ± 0.06** | 0.002 |
 1 (ms) 1 (ms) |
52.13 ± 15.91 | 49.67 ± 9.26 | 0.9 |
| c | 0.01 ± 0.01 | 0.01 ± 0.01 | 0.89 |
| n | 6 | 6 |
Alpha Pore Current Activation
Figure 1 shows original traces of representative calcium alpha pore currents recorded from GLT cells expressing WT (Figure 1A) or R528G (Figure 1B) mutant calcium channels. Both WT and R528G channels showed similar current density (Table 1). The parameters of the Boltzmann fits for steady-state activation revealed similar midpoints (V0.5=16.51±2.52mV for R528G vs. 20.25±1.94mV for WT) and similar steepness (k=7±1.94mV vs. 5.04±1.17 mV; Figure 1C). Therefore, the voltage-dependent activation of the mutant was comparable to wildtype. In contrast, there was an indication of a change of the kinetics of activation. Figure 1D shows the voltage dependence of τact of WT and R528G. Even though the voltage dependence of τact of the R528G currents had an overall similar trend to wildtype, significant differences in τact were present in voltages of +20mV to +50mV, indicating that the R528G mutation slowed the activation rate of the channel at more depolarized potentials.
Alpha Pore Current Inactivation
R528G showed a 13.65mV hyperpolarizing shift of the steady-state inactivation curve namely V0.5 = -36.98±4.71mV vs. -23.33±3.66mV (p=0.045), but there was no difference in the steepness between WT and R528G, with k= -12.59±0.71mV vs. -11.45±1.27mV (p=0.45; n=6 each). This facilitated inactivation suggested reduced channel function. Additionally, the time constant of inactivation τh was markedly faster in the mutant channel than in WT. In the WT channel, τh decreased steeply between +10mV and +20mV, a range over which the τh value of the mutant was nearly constant. The τh values of R528G were significantly smaller than the WT values at all measured voltages (Figure 1E, Table 1), namely 1.25±0.16ms vs. 2.75±0.37ms (10mV; p<0.0012), 0.70±0.07ms vs. 1.81±0.27ms (20mV; p<0.0019), 0.63±0.04ms vs. 1.59±0.14ms (30mV; p<0.000002) and 0.61±0.04ms vs. 1.37±0.15ms (40mV;p<0.00076) which also suggested a reduced alpha pore current of the mutant.
Alpha Pore Current Recovery from Inactivation
The mutant considerably altered the time course of channel recovery from inactivation at the recovery potentials of -70mV and -80mV (Figure 1F). The fractional amplitude A was lower in the mutant than in the WT channels indicating that the amount of recovering channels is significantly lower (0.61±0.02 vs. 0.74±0.02 at -80mV with p<0.003 for n=6; 0.42±0.06 vs. 0.69±0.02 at -70mV with p<0.002 for n=6; Figure 1F, Table 1). However, the time constant was not significantly altered at any of the holding potentials. The accelerated rate of inactivation and incomplete recovery from inactivation at -80mV and -70mV indicated a stabilization of the inactivated state of the mutant which was consistent with the left shift of the steady-state inactivation. Taken together, our data clearly demonstrated that the R528G mutation is a hypomorphic mutation and reduces the alpha pore current of the skeletal calcium channel.
Omega Currents
We found a hyperpolarization-induced inward current of the R528G mutant that was significantly higher than that of the wild-type channels and untransfected cells (Figure 2A) namely -2.57±0.28pA/pF vs. -1.59±0.27pA/pF (-140mV); -1.81±0.21pA/pF vs. -1.08±0.09pA/pF (-130mV); -1.32±0.2pA/pF vs. -0.73 ±0.16pA/pF (-120mV); -1.11±0.24pA/pF vs. -0.46±0.08pA/pF (-110mV) and -0.66±0.13pA/pF vs. -0.32±0.06pA/pF (-100mV) with p<0.05 and n=7. To characterize it properly, we subtracted the currents of untransfected cells (endogenous background currents) from mutant and WT currents. The resulting current for WT was linear and interpreted to be a background leak current, whereas the resulting current of the mutant was an exponentially decreasing voltage-dependent current comparable to previously described omega currents in a CaV1.1 HypoPP mutant 13. This omega current had a maximal conductance of 22.17nS/nF (Figure 2B).
Figure 2. Omega currents of CaV1.1 in GLT myotubes. Original traces of guanidinium omega currents from heterologously expressed (A) wild-type and (B) R528G channels and their calculated current densities (C) with additional untransfected GLT myotubes with usual guanidinium solution. (D) Plot of WT minus untransfected vs. mutant minus untransfected from C, fitted with a linear or exponential function. Dashed line indicates a maximal conductance of 22.17nS/nF of the mutant. All error bars represent the mean ± SEM. The p values are *<0.05.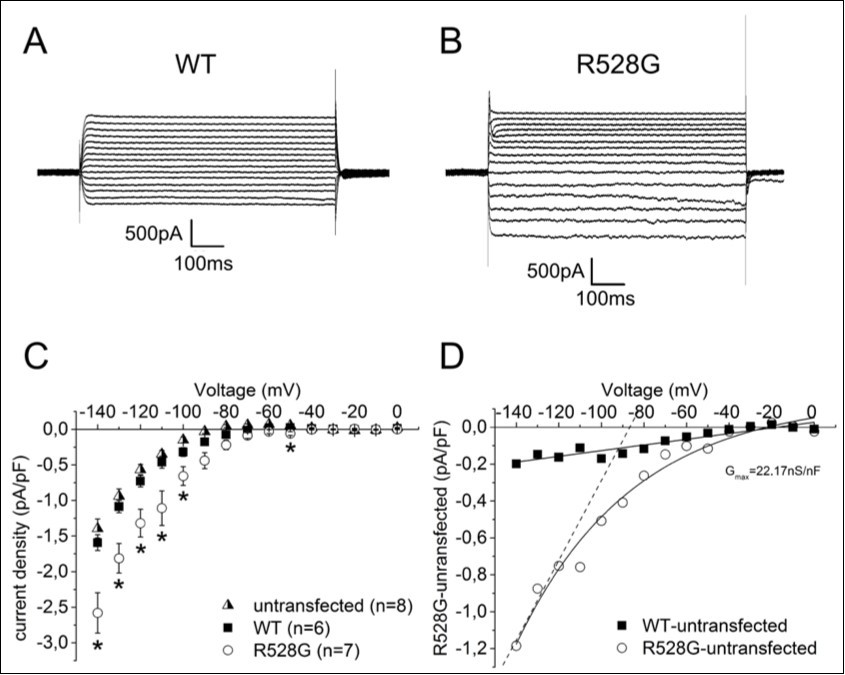
Download figure
Discussion
Comparison of Clinical Features of Cav1.1 R528 Mutants
Three CaV1.1 R528 mutations have been described clinically in the literature: R528H/G/C. An overview is given in Table 2, which includes the data of our patients 1, 5, 10, 11, 14, 15, 18, 19, 20. The age of onset for all mutations is approximately the 2nd decade, although the disease manifested slightly earlier in our R528G family. Contrary to expectations, R528G/C caused less frequent episodes of shorter duration than did R528H; however, in R528G patients ictal respiratory insufficiency 15 and cardiac involvement (our family) may imply greater severity of the weakness. Still, incomplete penetrance in women was observed for R528G (the proband’s mother and reference 14) just as in R528H 10. Unlike the R528H cases, no permanent weakness or vacuolar myopathy has been described in R528C/G patients, but this could be due to the small number of known patients. Therefore, although there is some support for more severe weakness, the phenotype of R528G cannot be clearly interpreted as more severe than the R528H phenotype.
Table 2. Clinical features of CaV1.1 R528 mutations.| R528H | R528G | R528C | ||
| our cases | literature | |||
| age at onset | 15 | 12 | 16.5 | 23 |
| frequency of episodes | weekly – monthly | yearly | weekly to monthly | monthly-yearly |
| duration of episodes | hours | minutes to hours | hours to months | minutes to hours |
| triggering factors | carbohydrate rich meals, rest after exercise | exercise or stress | carbohydrate rich meals, rest after exercise | exercise or stress |
| ictal K+ | 2mmol/l | 2.1mmol/l | 2.5mmol/l | 1.89mmol/l |
| therapy with acetazolamide | effective acetazolamide | acetazolamide with daily potassium | acetazolamide ineffective or not described | Not described |
| weakness | vacuolar myopathy,myopatic changes, proximal legs | weak in upper limbs, recurrent mild attacks | generalized, respiratory insufficiency | weak in right tights than in all limbs |
| permanent weakness | in 25% of patients | none | not described | not described |
| penetrance | reduced in women | incomplete | reduced in women | complete |
Triggering factors are similar for all mutations; however, the hypokalemia trigger is milder in R528G patients, which suggests that the omega current must be larger to be able to produce symptoms at higher potassium levels. In contrast to R528H patients for whom acetazolamide regularly ameliorates symptoms, such treatment for R528G patients was only beneficial when combined with daily potassium (our family), but not when administered alone as prophylactic measure 15. The previous studies suggesting that patients with the arginine-to-glycine mutation do not experience any benefit from acetazolamide 5 cannot be fully confirmed because of our family. However, the milder hypokalemic trigger observed in all cases would be in agreement with a larger underlying omega current of R528G compared with R528H.
Alpha Pore Currents
The slowing of the activation of R528G alpha pore currents is similar to that of the previously characterized mutant R528H 21. The fact that the activation became progressively slower relative to WT with increasing depolarization may indicate an increasing probability for a second open state. R528H showed such a second open state (called O2) that is more favored in the mutant than in the WT 22. Depending on the ion selectivity of O2, this state could result in an increased depolarization tendency and thereby inactivation of the action potential-generating Na+ channels. Additionally, a reduction in the Ca2+ current density or delay in the activation of the channel during activity might also disrupt the ability of calcium-activated potassium channels to rescue the muscle cell after a series of action potentials, leading to depolarization and failure of conduction 21 thus contributing to the weakness.
The left shift of the steady-state inactivation is a recurrent finding for CaV1.1 mutations, i.e., R1242G 13 and R528H 16. Combined with accelerated inactivation and incomplete recovery in R528G, this finding indicates a stabilization of the fast inactivated state and an overall reduced alpha pore current. This would be compatible with permanent weakness which surprisingly was not found clinically. The question arises whether the stabilized inactivated stated limits the duration of weakness episodes because the S4 segment is moved outward in the inactivated state which would shift the residue 528 outside of the gating pore constriction thereby terminating the omega current.
Omega Currents
Our voltage-clamp measurements of R528G channels expressed in GLT cells revealed a voltage-dependent inward current activated by hyperpolarization with the maximal conductance of 22.17nS/nF in our GLT expression system. Using the same method in form of a linear fit through the inward current at the voltages between -140mV and -120mV, homozygous CaV1.1 R528Hm/m fibers had maximal omega currents of 28nS/nF 12. Because of the different expression systems used, we cannot directly conclude whether the omega current of R528G is actually larger or smaller than that of R528H, however the general magnitude range is similar.
Conclusion
Our data did not confirm the three starting hypotheses because i) the phenotype was not clearly more severe for R528G than for R528H, ii) acetazolamide ameliorated symptoms in combination with daily potassium administration and iii) the omega current for R528G was not larger than for R528H. However, our data confirms that a large omega current is present in the resting potential range which is in agreement with current pathogenetic models of HypoPP 8.
References
- 1.Cavel-Greant D, Lehmann-Horn F, Jurkat-Rott K. (2012) The impact of permanent muscle weakness on quality of life in periodic paralysis: a survey of 66 patients. , Acta Myol 31(2), 126-33.
- 3.Suetterlin K, Männikkö R, Hanna M G. (2014) Muscle channelopathies: recent advances in genetics, pathophysiology and therapy. , Curr Opin Neurol 27(5), 583-90.
- 4.Catterall W A. (2010) Signaling complexes of voltage-gated sodium and calcium channels. , Neurosci Lett 486(2), 107-16.
- 5.Matthews E, Portaro S, Ke Q, Sud R, Haworth A. (2011) Acetazolamide efficacy in hypokalemic periodic paralysis and the predictive role of genotype. , Neurology 77(22), 1960-4.
- 6.Sokolov S, Scheuer T, Catterall W A. (2007) Gating pore current in an inherited ion channelopathy. , Nature 446(7131), 76-8.
- 7.Struyk A F, Cannon S C. (2007) A Na+ channel mutation linked to hypokalemic periodic paralysis exposes a proton-selective gating pore. , J Gen Physiol 130(1), 11-20.
- 8.Jurkat-Rott K, Groome J, Lehmann-Horn F. (2012) Pathophysiological role of omega pore current in channelopathies. , Front Pharmacol 3, 112.
- 9.Matthews E, Labrum R, Sweeney M G, Sud R, Haworth A. (2009) Voltage sensor charge loss accounts for most cases of hypokalemic periodic paralysis. , Neurology 72(18), 1544-7.
- 10.Elbaz A, Vale-Santos J, Jurkat-Rott K, Lapie P, Ophoff R A. (1995) Hypokalemic periodic paralysis and the dihydropyridine receptor (CACNL1A3): genotype/phenotype correlations for two predominant mutations and evidence for the absence of a founder effect in 16 caucasian families. , Am J Hum Genet 56(2), 374-80.
- 11.Jurkat-Rott K, Weber M-A, Fauler M, Guo X-H, Holzherr B D. (2009) K+-dependent paradoxical membrane depolarization and Na+ overload, major and reversible contributors to weakness by ion channel leaks. Proc Natl Acad Sci U S A 106(10), 4036-41.
- 12.Wu F, Mi W, Hernández-Ochoa E O, Burns D K, Fu Y. (2012) A calcium channel mutant mouse model of hypokalemic periodic paralysis. , J Clin Invest 122(12), 4580-91.
- 13.Fan C, Lehmann-Horn F, Weber M-A, Bednarz M, Groome J R. (2013) Transient compartment-like syndrome and normokalaemic periodic paralysis due to a Ca(v)1.1 mutation. Brain. 136(Pt 12): 3775-86.
- 14.Wang Q, Liu M, Xu C, Tang Z, Liao Y. (2005) Novel CACNA1S mutation causes autosomal dominant hypokalemic periodic paralysis in a Chinese family. , J Mol Med (Berl) 83(3), 203-8.
- 15.Kil T-H, Kim J-B. (2010) Severe respiratory phenotype caused by a de novo Arg528Gly mutation in the CACNA1S gene in a patient with hypokalemic periodic paralysis. , Eur J Paediatr Neurol 14(3), 278-81.
- 16.Jurkat-Rott K, Uetz U, Pika-Hartlaub U, Powell J, Fontaine B. (1998) Calcium currents and transients of native and heterologously expressed mutant skeletal muscle DHP receptor alpha1 subunits (R528H). , FEBS Lett 423(2), 198-204.
- 17.Powell J A, Petherbridge L, Flucher B E. (1996) Formation of triads without the dihydropyridine receptor alpha subunits in cell lines from dysgenic skeletal muscle. , J Cell Biol 134(2), 375-87.
- 18.Yang B, Yang Y, Tu W, Shen Y, Dong Q. (2014) A rare case of unilateral adrenal hyperplasia accompanied by hypokalaemic periodic paralysis caused by a novel dominant mutation in CACNA1S: features and prognosis after adrenalectomy. , BMC Urol 14, 96.
- 19.Sternberg D, Maisonobe T, Jurkat-Rott K, Nicole S, Launay E. (2001) Hypokalaemic periodic paralysis type 2 caused by mutations at codon 672 in the muscle sodium channel gene SCN4A.Brain.124(Pt 6):. 1091-9.
- 20.Miller T M, MR Dias da Silva, Miller H A, Kwiecinski H, Mendell J R. (2004) Correlating phenotype and genotype in the periodic paralyses. , Neurology 63(9), 1647-55.
Cited by (3)
This article has been cited by 3 scholarly works according to:
Citing Articles:
The Journal of General Physiology (2018) OpenAlex Semantic Scholar Crossref
The Journal of General Physiology (2018) OpenAlex
Fenfen Wu, M. Quiñonez, M. DiFranco, S. Cannon - The Journal of General Physiology (2018) Semantic Scholar Crossref