
Cloning, Expression and Characterization of the α-glucuronidase from the Hyperthermophile DictyoglomusturgidumDSM 6724Ô
Abstract
Conversion of biomass into fermentable sugars is a major requirement for successful and cost-effective biofuels production. The conversion of xylan to sugars requires multiple enzymes including α-glucuronidase. Here we report the cloning, expression, purification and characterization of the α-glucuronidase from Dictyoglomusturgidum(DtuAgu). DtuAgu is an intracellular protein of 685 amino acids and a predicted molecular weight of 79.4 kD. Enzymatic activity was optimum between pH 7.0 and 8.0 and at 85°C. The specific activity of the enzyme was 10 u/mg when measured using mixed aldouronic acids. The specific activity on isolated glucuronoxylan was approximately 20% of the value obtained with xylooligosaccharides. DtuAgu significantly improved xylan conversion to xylose when evaluated using two mixtures of thermostable bacterial enzymes and two sources of xylan. DtuAgu has the potential to be a key player in thermostable enzyme cocktails for the conversion to biomass to biofuels.α
Article Information
- Received
- Accepted
- Published
Academic Editor: Mezni Ali, Department of Life Sciences, University of Carthag Tunisia.
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2020 Phillip Brumm, et al.
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Corresponding author: Phillip Brumm, C5-6 Technologies, 5627 Old Oak Drive, Fitchburg, WI 53711 —
Competing Interests
PB is founder and CEO of C5-6 Technologies LLC, a company started to make the biomass-degrading enzymes developed by the GLBRC available to researchers at a nominal charge. The company was formed after the completion of the work presented here, and was not involved in the study design, collection, analysis and interpretation of data.
Funding
No specific funding statement was provided by the authors.
Data Availability
No data-availability statement was provided by the authors.
Acknowledgements
This work was funded by the DOE Great Lakes Bioenergy Research Center (DOE BER Office of Science DE-FC02-07ER64494). The funding agency was not involved in the study design, collection, analysis and interpretation of data; or in the writing of the manuscript.
Citation:
Introduction
Plant-based biomass is made up of two main polysaccharide components, cellulose and hemicellulose. Conversion of these polysaccharides to bio-based fuels and chemicals requires the degradation of the cellulose and hemicellulose to sugar monomers via enzymatic processes. A number of enzymatic systems have been described for achieving these conversions, including systems from bacterial 1 and fungal 2, 3, 4, 5 sources. The degradation of these polysaccharides is made more difficult by the complexity of the substrates. While cellulose is a homopolymer of β-1,4 linked glucose, xylans are a heterogenous collection of molecules sharing only a β-1,4 linked xylose backbone. Arabinoxylans contain xylose residues highly substituted with α-(1,2)-linked arabinose, α-(1,3)- linked arabinose as well as xylose residues substituted with both α-(1,2), α-(1,3)-linked arabinose, with a ratio of arabinose to xylose of 0.5 to 0.6 for wheat arabinoxylan 6. Glucuroxylans, found in hardwoods, contain α-(1,2)-linked D-glucuronic acid and 4-O-methyl-D-glucuronic acid residues as well as acetic acid. These D-glucuronic acid and 4-O-methyl-D-glucuronic acid residues are attached to roughly every tenth xylosyl residue in the xylan backbone. Arabinoglucuronoxylans, found in softwoods, contain both α-(1,2)-4-O-methyl-D-glucuronic acid and α-(1,3)-linked arabinose residues.
Enzymatic degradation of xylans requires the participation of a number of enzymes to completely convert the xylans to monosaccharides. α-Glucuronidases, α-arabinofuranoidases, and esterases are needed to remove the sidechains from the xylans, and xylanases and xylosidases are needed to convert the backbone to xylose. α-Glucuronidases (EC 3.2.1.139) hydrolyze the α-1,2 glyosidic bond between α-D-glucuronic acid (GlcA) or its 4-O-methyl ether (MeGlcA) and xylose residues of xylooligosaccharides. Structurally, α-glucuronidases are found in glycoside hydrolase families 67 (GH67) 7, 8, 9, 10, 11 and 115 (GH115) 12, 13, 14, 15, 16. These enzymes are much more efficient in removing uronic acid from glucuronoxylooligosaccharides than from native glucuronoxylan or arabinoglucuronoxylan 17. The structures of only two GH67 α-glucuronidases, from Geobacillus stearothermophilus18 and Cellvibrio japonicus Ueda107 7 have been determined and published. The limited number of α-glucuronidases that are available suggest that additional characterized GH67 and GH115 family members may assist with developing effective means of degrading xylan. High temperature bioprocesses have numerous advantages over their mesophilic counterparts 19. Development of new and highly effective enzymes including α-glucuronidases that operate at high temperature will speed up bioprocess development.
Dictyoglomusspecies are genetically distinct organisms that have been identified in anaerobic, hyperthermophilic hot spring environments 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31. There are only two validly described Dictyoglomus strains, Dictyoglomusthermophilum which was isolated from Tsuetate Hot Spring in Kumamoto Prefecture, Japan 32.The genome of D. thermophilum has been sequenced 31, and a number of potentially useful enzymes including amylases 33, 34, xylanases 35, 36, a mannanase 37 and an endoglucanase 38 have been cloned and characterized. The second described species, Dictyoglomusturgidus, was isolated from a hot spring in the Uzon Caldera, in eastern Kamchatka, Russia 30. The name Dictyoglomusturgidus was subsequently corrected to Dictyoglomusturgidum39. Enzyme library construction and carbohydrase screening was performed using D. turgidum genomic DNA 40, as well as whole genome sequencing 41. Preliminary results on the cloning and successful evaluation of the D. turgidumα-glucuronidase (DtuAgu) have been reported 42; here we present the detailed characterization of the enzyme.
Material and Methods
Materials
D. turgidum strain 6724 T was obtained from the Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (DSMZ). 10G electrocompetent E. coli cells and pEZSeq (a lac promoter vector), were obtained from Lucigen, Middleton, WI. Wheat arabinoxylan (low viscosity) and α-D-glucuronidase test kit K-AGLUA were obtained from Megazyme International (Wicklow, Ireland). Polyacrylamide electrophoresis gels were obtained from Bio-Rad, Hercules, CA and Bullseye Pre-stained Protein ladder was obtained from Midwest Scientific (Valley Park, MO). Pierce™ Coomassie Plus (Bradford) Assay Kit was obtained from Thermo-Fisher (Waltham, MA). Beechwood 4-O-Methyl-D-glucurono-D-xylan (glucuronoxylan), vancomycin, ampicillin, Vivaspin 20 ultrafiltration units with 10,000 mwco membranes, Sephacryl S-400 High Resolution and Gel Filtration Markers Kit for Protein Molecular Weights 12,000-200,000 Daltons were obtained from Millipore Sigma, St. Louis, MO. Ammonia fiber expansion (AFEX)-treated corn stover was obtained from the Great Lakes Bioenergy Research Center (GLBRC), Madison, WI. Secreted Geobacillus xylanase P005 (G11MC16DRAFT_1587), intracellular Geobacillus xylanase P006 (Y412MC52_1840), Geobacillus α-arabinofuranosidase (Ara1) (P031, G11MC16DRAFT_1557), and Geobacillus xylosidase (Xyl1) (P046, Y412MC61_2711) were obtained from C5-6 Technologies LLC (Fitchburg, WI).
Methods
D. turgidum DSM 6724 TM was grown and a genomic library prepared as described previously 41, 43 YT plate media (16 g/l tryptone, 10 g/l yeast extract, 5 g/l NaCl and 16 g/l agar) was used in all molecular biology screening experiments. Terrific Broth (12 g/l tryptone, 24 g/l yeast extract, 9.4 g/l K2HPO4, 2.2 g/l KH2PO4, and 4.0 g/l glycerol added after autoclaving) was used for liquid cultures.
The genomic DNA sequence was converted to protein sequence using the Expasy Translate tool https://web.expasy.org/translate/?_ga=1.260305485.1997906226.1473781687.
InterProScan Family analysis (http://www.ebi.ac.uk/Tools/InterProScan/ ), and BLASTP (Basic Local Alignment Search Tool 44 (http://blast.ncbi.nlm.nih.gov/Blast.cgi) analysis tools were used to compare DtuAgu with other proteins in the database. Phylogeny analysis was performed using software at http://www.phylogeny.fr/version2_cgi/index.cgi.
Sequence alignment of D. turgidum, T. maritima, and G.stearothermophilus α-glucuronidase sequences was performed using T-Coffee software at http://tcoffee.crg.cat/apps/tcoffee/result?rid=328b2609 45, 46, 47. For tree construction, multiple alignments were run using ClustalW 48 alignment curation was done to remove positions with gaps 49, construction of the phylogenetic tree was done using PhyML 50, and visualization was done using TreeDyn 51. Determination of the presence of absence of a signal peptide was performed using SignalP 52. Three-dimensional structure prediction was performed using SWISS-MODEL (https://swissmodel.expasy.org ).
The α-glucuronidase gene was amplified, ligated into pET28A, and transformed into BL21(DE3) E. coli competent cells. Recombinant clones were cultured overnight at 37°C, 100 rpm, in 100 ml Luria Broth containing 50 mg/l kanamycin. Expression was induced using 1 mM IPTG, and cultures were harvested 18 h after induction. Cells were pelleted by centrifugation, and the pellets were lysed by sonication. Proteins were purified using standard methods for His-tagged proteins 53. Protein purity and approximate molecular weight were determined by SDS PAGE on Bio-Rad 4% to 20% acrylamide gels using the Bullseye Pre-stained Protein ladder. Protein concentration was determined using the Coomassie Plus (Bradford) Assay Kit using bovine serum albumin as standard. Solution molecular weight was determined by gel filtration using a 2.5cm x 70 cm Sephacryl S-400 HR column equilibrated with 100 mM Tris-HCl pH 8.0 containing 250 mM NaCl calibrated with the Millipore Sigma Gel Filtration Markers Kit.
The activity of the purified enzyme was determined using the Megazyme K-AGLUA assay kit manual method. Enzyme was incubated in 1% solution of NaBH4-reduced Mixed Aldouronic Acids (Tri:Tetra:Penta = 2:2:1; Megazyme, O-AMXR) in 50 mM sodium acetate, pH 5.8 at 70°C for 20 minutes, followed by measurement of the formed glucuronic acid using the supplied method. Measurement of the enzyme activity with 4-O-Methyl-D-glucurono-D-xylan was conducted as described above, except a 2 g/l solution of 4-O-Methyl-D-glucurono-D-xylan in 50 mM sodium acetate, pH 5.8. One α-glucuronidase unit will produce 1 micromole of reducing sugar per minute at 70°C and pH 5.8. Enzyme activities were measured in triplicate using at least two separate enzyme samples. For polysaccharide hydrolysis experiments, monosaccharide release was determined using 1000 ml of 0.2% polysaccharide in 100 mM acetate buffer, pH 5.8 and 50°C. In addition, all long-term reactions contained 5mg/ml vancomycin and 10mg/ml ampicillin to prevent microbial growth. Enzyme dosing unless noted elsewhere was 20 mg/ml of pure enzyme. Aliquots of the reaction mixture were removed, the reaction was stopped by incubation at 95°C, and xylose production was measured using the Megazyme K-XYLOSE xylose kit using the manufacturer’s instructions. All assay values are the average of triplicate measurements performed on at least two samples.
Results
The protein sequence of DtuAgu was translated from the genomic DNA sequence (Figure 1) corresponding to gene Dtur_1714. The protein sequence is available as UniProtKB - B8E3B2 (B8E3B2_DICTD) and NCBI Reference Sequence: WP_012584061.1.
Figure 1. DtuAgu Protein Sequence (Dtur_1714)
Download figure
Phylogenetic analysis was conducted using the software package Phylogeny.fr with the most closely related α-glucuronidase sequences identified by BLAST analysis. The secreted, fungal Trichoderma reesei α-glucuronidase was used to root the tree (Figure 2). DtuAgu is highlighted in red, while the only two other characterized thermostable α-glucuronidases, G. stearothermophilus and T. maritima α-glucuronidases, are highlighted in blue.
Figure 2. Phylogenetic Tree of α-Glucuronidases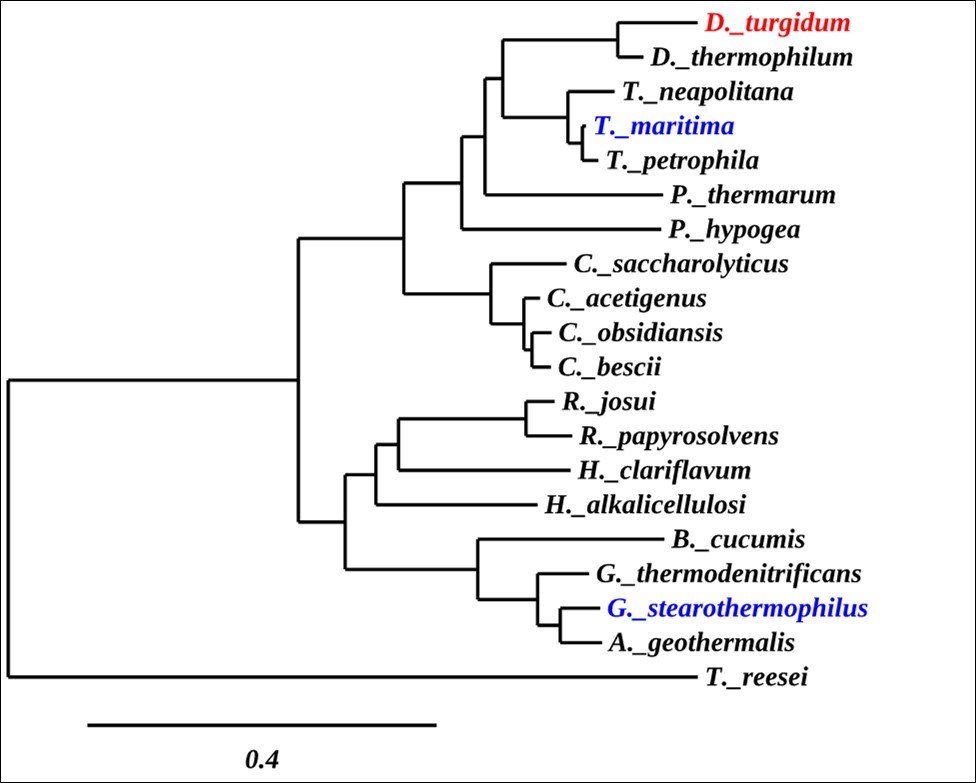
Download figure
The phylogenetic analysis shows that DtuAgu is closely related to two other Dictyoglomusα-glucuronidases. The next closest relatives of DtuAgu are α-glucuronidases from two families of the thermophilic, gram-negative phylum Thermotogae, Thermatogaceae and Petrotogaceae. DtuAgu is also closely related to members of the genus Caldicellulosiruptor, a group of organisms previously claded with Thermoanaerobacterium. None of the Caldicellulosiruptor α-glucuronidases have been cloned and characterized. DtuAgu is only distantly related to the well-characterized, α-glucuronidase of the gram-positive G.stearothermophilus (GstAgu) and other Geobacillusspecies. Sequence Alignment of DtuAgu with the characterized GstAgu and T. maritima α-glucuronidase (TmarAgu) using T-Coffee software identifies 388/687 amino acid identities between GstAgu and DtuAgu and 475/687 amino acid identities between DtuAgu and TmarAgu. The sequence matches are not evenly distributed throughout the three proteins, with most of the sequence divergence occurring within the first 190 amino acids of the proteins (Figure 3).
Figure 3. Sequence alignment of DtuAgu, TmarAgu and GstAgu
Download figure
Based on SignalP analysis, the enzyme did not possess a signal peptide and is most likely an intracellular enzyme.
The gene encoding DtuAgu was cloned into E. coli with a N-terminal 6-His affinity tag and expressed. After cell lysis and clarification, the enzyme was purified using a single step of immobilized metal affinity chromatography (IMAC) to >95% purity (Figure 4). Based on the amino acid sequence of DtuAgu, the predicted molecular weight of the protein is 79,447 daltons, slightly higher than the value predicted by the mobility on SDS PAGE.
Figure 4. SDS PAGE of Purified DtuAgu
Download figure
DtuAgu had a temperature optimum of 85°C when assayed at pH 7.5 (Figure 5), and a pH optimum between pH 7.0 and 8.0 at 70°C (Figure 6).
Figure 5. Temperature-activity Relationship of DtuAgu
Download figure
Figure 6. pH-activity Relationship of DtuAgu
Download figure
When the assayed at 70°C, pH 7.5, the specific activity of DtuAgu was 10.0 ± 1.0 U/mg using the Megazyme Mixed Aldouronic Acids. When assayed at 70°C, pH 5.8, conditions identical to those used by our group for biomass hydrolysis using thermophilic cellulases and xylanases, DtuAgu had a specific activity of 2.0 ± 0.2 U/mg when assayed using the same substrate. The specific activity was highly reproducible when assayed using a single lot of Megazyme substrate, but different substrate lots gave values differing by as much as 30%. Unlike other α-glucuronidases, DtuAgu had detectable activity on intact xylan, with a specific activity of 0.4 ± 0.1 U/mg at 70°C, pH 5.8, using glucuronoxylan.
To better understand the activity of the enzyme in the presence of other xylan-degrading enzymes, α-glucuronidase activity was assayed by measuring percent of xylose released from substrates using control mixtures of xylanase, xylosidase and α-arabinofuranosidase. The specificity of DtuAgu was first evaluated at pH 5.8 and 60°C using low viscosity wheat arabinoxylan, a substrate that contains no MeGlcA. As expected, the results (Table 1) show DtuAgu does not yield a statistically significant increase in xylose production when evaluated with either P006 and P031, or P006, P031, and P046. Xylose conversions of >100% are the result of increased recovery of xylose from enzymatic hydrolysis versus acid hydrolysis.
Table 1. Arabinoxylan Hydrolysis| Enzyme Mixture | Conversion, 20 hr | Conversion, 44 hr |
| P006 + P031 | 59% | 59% |
| P006 + P031 + DtuAgu | 63% | 60% |
| P006 + P031 + P046 | 106% | 104% |
| P006 + P031 + P046+ DtuAgu | 104% | 112% |
DtuAgu was then evaluated using two substrates containing MeGlcA, glucuronoxylan and AFEX-treated corn stover. Glucuronoxylan was hydrolyzed at pH 5.8 and 60°C using either P006, P031, and P046 (Control 1) P005, P031, and P046 (Control 2) with and without supplemental DtuAgu. Hydrolysis of glucuronoxylan from beechwood was conducted as described in Materials and Methods. The predominant sidechain substitution in beechwood xylan is MeGlcA, 13% of the polymer weight. Arabinose and acetyl groups are present in only very low levels in this substrate. DtuAgu significantly improves the production of xylose from this substrate, increasing xylose yield 22% to 28% with the Control 1 enzymes, and 30% to 32% with the Control 2 enzymes. Figure 7.
Figure 7. Glucuronoxylan conversion
Download figure
The experiment was repeated using the same enzymes and reaction conditions, with AFEX-treated corn fiber. The corn fiber xylan contains significantly less MeGlcA and more acetyl and arabinose substitution than beechwood xylan. Again, DtuAgu increased xylose yield 4% to 7% with the Control 1 enzymes, and 12% to 13% with the Control 2 enzymes. Figure 8.
Figure 8. AFEX Corn Stover conversion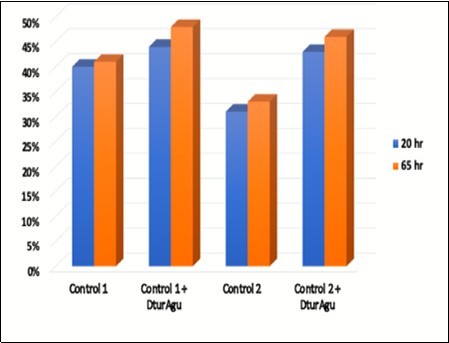
Download figure
Discussion
In most proposed processes the conversion of biomass to fuels requires first the pretreatment of biomass to remove lignin and improve enzymatic digestibility, second, the conversion of the pretreated mass to monosaccharides, and finally, the fermentation of the monosaccharides to fuels and chemicals 54. The complexity of the cellulose and hemicellulose components found in the biomass requires many individual enzymes to achieve high-level conversion. The cellulose-degrading enzymes needed include endo-acting and exo-acting cellulases and β-glucosidases, and the hemicellulose-degrading enzymes needed include xylanases, β-xylosidases, α-arabinofuranosidases, xylan esterases, and α-glucuronidases. Effective conversion of just the cellulose component still requires numerous hemicellulose-degrading enzymes to achieve this goal 42. Further complicating the problem of biomass degradation is that several of the enzymes including β-glucosidases, β-xylosidases, α-arabinofuranosidases, xylan esterases, and α-glucuronidases are normally intracellular enzymes and must be prepared separately and added to the cocktail of secreted enzymes. Finally, all enzymes in the final cocktail must have similar pH and temperature optima that allow them to work under the same reaction conditions.
This work describes the cloning, purification and characterization of the α-glucuronidase from the hyperthermophile, D. turgidum. The purified DtuAgu is a 79.4 kD protein, similar in size to TmarAgu, 76.2 kD, and GstAgu, 78.4 kD. The gene encoding DtuAgu is most closely related to α-glucuronidase genes of other Dictyoglomus species, followed by Thermotoga and Caldicellulosiruptorspecies. Alignment of the DtuAgu, TmarAgu, and GstAgu genes show a divergent, approximately 200 amino acid N-terminal region of the protein; the remainder of the proteins are highly conserved in sequence. The differences in the N-terminal sequences may be responsible for differences in the native forms of the enzymes. In this work, DtuAgu was found to be monomeric in solution, while TmarAgu was reported to be multimeric 55 and GstAgu was reported to be dimeric 18, 56. The temperature optimum of DtuAgu is 85°C, identical to that reported for TmarAgu 55 , and significantly higher than the 65°C temperature optimum for GstAgu 56. The pH/activity curve of DtuAgu is similar to that reported for TmarAgu 55. The specific activity was 10 u/mg protein/ lower than the reported value of 31 u/mg reported for TmarAgu measured using a different substrate no longer available. DtuAgu was active on intact glucuronoxylan, with a specific activity of approximately 20% of that measured using xylan oligosaccharides. This is the first report of thermostable α-glucuronidase activity on intact xylan. Previous work by our group demonstrated the ability of DtuAgu to improve performance of fungal Trichoderma reesei enzymes in biomass conversion 42. Here we demonstrate the ability of the DtuAgu to also significantly improve performance of bacterial xylanolytic enzymes in conversion of isolated xylan and pretreated biomass.
Conclusions
DtuAgu has potential for improving the conversion and reducing the cost of biomass conversion into fermentable sugars, a major requirement for cost-effective biofuel production. The extent of improvement in xylose by DtuAgu cannot be accurately predicted because it depends strongly on the experimental conditions. Large differences in xylose yield were seen between different sources of xylan hydrolyzed with the same enzyme cocktail, and large differences were also seen between the same substrate hydrolyzed with different xylanases. Additional work is needed to evaluate and optimize enzyme cocktails for biomass conversion, possibly optimizing individual cocktails for each biomass source.
Authors Contributions
PB designed the study, analyzed the data, purified the enzymes and wrote the first draft of the manuscript. DX performed all cellulose degradation studies. LA cloned DtuAgu. DM managed the enzyme cloning and expression, as well as all DNA sequencing used in the cloning. All authors read and approved the final manuscript.
References
- 1.P J Brumm. (2013) Bacterial genomes: what they teach us about cellulose degradation.Biofuels4. 669-681.
- 2.Peterson R, Nevalainen H. (2012) Trichoderma reesei RUT-C30--thirty years of strain improvement.Microbiology158. 58-68.
- 3.Li Y, Liu C, Bai F, Zhao X. (2016) Overproduction of cellulase byTrichodermareeseiRUT C30 through batch-feeding of synthesized low-cost sugar mixture.BioresourTechnol216. 503-510.
- 4.Wang S, Liu G, Wang J, Yu J, Huang B et al. (2013) Enhancing cellulase production inTrichodermareeseiRUT C30 through combined manipulation of activating and repressing genes.J. IndMicrobiolBiotechnol40 633-641.
- 5.Li Z, Yao G, Wu R, Gao L, Kan Q et al. (2015) Synergistic and Dose-Controlled Regulation of Cellulase Gene Expression inPenicilliumoxalicum.PLoSgenetics11. 1005509.
- 6.Kosik O, S J Powers, Chatzifragkou A, P C, Charalampopoulos D et al. (2017) Changes in the arabinoxylan fraction of wheat grain during alcohol production.Food Chem221. 1754-1762.
- 7.Nurizzo D, Nagy T, H J Gilbert, G J Davies. (2002) The structural basis for catalysis and specificity of thePseudomonascellulosaalpha-glucuronidase. GlcA67A.Structure10 547-556.
- 8.Shulami S, Gat O, A L Sonenshein, Shoham Y. (1999) The glucuronic acid utilization gene cluster fromBacillus stearothermophilusT-6.JBacteriol181. 3695-3704.
- 9.Nagy T, Emami K, C M Fontes, L M Ferreira, D R Humphry et al. (2002) The membrane-bound alpha-glucuronidase fromPseudomonascellulosahydrolyzes 4-O-methyl-D-glucuronoxylooligosaccharides but not 4-O-methyl-D-glucuronoxylan.JBacteriol184. 4925-4929.
- 10.Jia X, Mi S, Wang J, Qiao W, Peng X et al. (2014) Insight into glycoside hydrolases for debranched xylan degradation from extremely thermophilic bacteriumCaldicellulosiruptorlactoaceticus.PLoSOne9. 106482.
- 11.Y H Moon, Iakiviak M, Bauer S, R I Mackie, I K Cann. (2011) Biochemical analyses of multiple endoxylanases from the rumen bacteriumRuminococcusalbus8 and their synergistic activities with accessory hemicellulose-degrading enzymes.ApplEnvironMicrobiol77. 5157-5169.
- 12.Rogowski A, Basle A, C S Farinas, Solovyova A, J C Mortimer et al. (2014) Evidence that GH115alpha-glucuronidase activity, which is required to degrade plant biomass, is dependent on conformational flexibility.JBiolChem289. 53-64.
- 13.S L Chong, Derba-Maceluch M, Koutaniemi S, L D Gomez, S J McQueen-Mason et al. (2015) Active fungal GH115alpha-glucuronidase produced inArabidopsis thalianaaffects only the UX1-reactive glucuronate decorations on native glucuronoxylans.BMCBiotechnol15. 56.
- 14.Wang W, Yan R, B P Nocek, T V Vuong, R Di Leo et al. (2016) Biochemical and Structural Characterization of a Five-domain GH115alpha-Glucuronidase from the Marine BacteriumSaccharophagusdegradans2-40T.JBiolChem291. 14120-14133.
- 15.Fujimoto Z, Ichinose H, Biely P, Kaneko S. (2011) Crystallization and preliminary crystallographic analysis of the glycoside hydrolase family 115 alpha-glucuronidase fromStreptomycespristinaespiralis.ActaCrystallogrSect F StructBiolCrystCommun67. 68-71.
- 16.L S McKee, Sunner H, G E Anasontzis, Toriz G, Gatenholm P et al. (2016) A GH115 alpha-glucuronidase fromSchizophyllumcommunecontributes to the synergistic enzymatic deconstruction of softwood glucuronoarabinoxylan.BiotechnolBiofuels9. 2.
- 17.D C Smith, C W Forsberg. (1991) . alpha-Glucuronidase and Other Hemicellulase Activities ofFibrobactersuccinogenesS85 Grown on Crystalline Cellulose or Ball-Milled Barley Straw.ApplEnvironMicrobiol57 3552-3557.
- 18.Golan G, Shallom D, Teplitsky A, Zaide G, Shulami S et al. (2004) Crystal structures ofGeobacillusstearothermophilus alpha-glucuronidase complexed with its substrate and products: mechanistic implications.JBiolChem279. 3014-3024.
- 19.B M Zeldes, M W Keller, A J Loder, C T Straub, M W Adams et al. (2015) Extremely thermophilic microorganisms as meFigolic engineering platforms for production of fuels and industrial chemicals.FrontMicrobiol6. 1209.
- 20.Mathrani I, Ahring B. (1991) Isolation and characterization of a strictly xylan-degradingDictyoglomusfrom a man-made, thermophilic anaerobic environment.ArchMicrobiol157. 13-17.
- 21.Mathrani I, Ahring B. (1992) Thermophilic and alkalophilic xylanases from severalDictyoglomusisolates.ApplMicrobiolBiotechnol38. 23-27.
- 22.B K Patel, H W Morgan, Wiegel J, R M Daniel. (1987) Isolation of an extremely thermophilic chemoorganotrophic anaerobe similar toDictyoglomusthermophilumfrom new zealand hot springs.ArchMicrobiol147. 21-24.
- 23.E A Burgess, J M Unrine, G L Mills, C S Romanek, Wiegel J. (2012) Comparative geochemical and microbiological characterization of two thermal pools in the Uzon. , Caldera, Kamchatka, Russia.MicrobEcol63 471-489.
- 24.I V Kublanov, A, G B Slobodkina, A V Lebedinsky, S K Bidzhieva et al. (2009) Biodiversity of thermophilic prokaryotes with hydrolytic activities in hot springs of Uzon Caldera. , Kamchatka (Russia).ApplEnvironMicrobiol75: 286-291.
- 25.V M Gumerov, A V Mardanov, A V Beletskii, E A Bonch-Osmolovskaia, N V Ravin. (2011) [Molecular analysis of microbial diversity in the Zavarzin Spring, the Uzon caldera ].Mikrobiologiia80. 258-265.
- 26.T V Kochetkova, Rusanov Pimenov, V N, T V Kolganova, A V Lebedinsky et al. (2011) Anaerobic transformation of carbon monoxide by microbial communities of Kamchatka hot springs.Extremophiles15. 319-325.
- 27.Sahm K, John P, Nacke H, Wemheuer B, Grote R et al. (2013) High abundance of heterotrophic prokaryotes in hydrothermal springs of the Azores as revealed by a network of 16S rRNA gene-based methods.Extremophiles17. 649-662.
- 28.Menzel P, S R Gudbergsdottir, A G Rike, Lin L, Zhang Q et al. (2015) Comparative Metagenomics of Eight Geographically Remote Terrestrial Hot Springs.MicrobEcol70. 411-424.
- 29.T A Vishnivetskaya, S D Hamilton-Brehm, Podar M, J, A V Palumbo et al. (2015) Community analysis of plant biomass-degrading microorganisms from Obsidian Pool, Yellowstone National Park.MicrobEcol69. 333-345.
- 30.Svetlichnii V, Svetlichnaya T. (1988) nov., a new extremely thermophilic eubacterium isolated from hot springs of the Uzon volcano caldera.Mikrobiologiya57. 364-369.
- 31.D A Coil, J H Badger, H C Forberger, Riggs F, Madupu R et al. (2014) Complete Genome Sequence of the Extreme ThermophileDictyoglomusthermophilumH-6-12.GenomeAnnounc2.
- 32.Saiki T, Kobayashi Y, Kawagoe K, Beppu T. (1985) Dictyoglomus thermophilum gen. nov., sp. nov., a Chemoorganotrophic, Anaerobic, Thermophilic Bacterium.Int JSystEvolMicrobiol35. 253-259.
- 33.Fukusumi S, Kamizono A, Horinouchi S, Beppu T. (1988) Cloning and nucleotide sequence of a heat-stable amylase gene from an anaerobic thermophile. , Dictyoglomusthermophilum.Eur 174, 15-21.
- 34.Horinouchi S, Fukusumi S, Ohshima T, Beppu T. (1988) Cloning and expression in Escherichia coli of two additional amylase genes of a strictly anaerobic thermophile,Dictyoglomusthermophilum,and their nucleotide sequences with extremely low guanine-plus-cytosine contents.Eur JBiochem176. 243-253.
- 35.M D Gibbs, R A, P L Bergquist. (1995) Cloning, sequencing, and expression of a xylanase gene from the extreme thermophileDictyoglomusthermophilumRt46B.1 and activity of the enzyme on fiber-bound substrate.ApplEnvironMicrobiol61. 4403-4408.
- 36.D, M D Gibbs, C W, M H Koh, K et al. (1998) Cloning of the xynB gene fromDictyoglomusthermophilumRt46B.1 and action of the gene product on kraft pulp.ApplEnvironMicrobiol64. 1759-1765.
- 37.M D Gibbs, R A, Sunna A, P L Bergquist. (1999) Sequencing and expression of a beta-mannanase gene from the extreme thermophileDictyoglomusthermophilumRt46B.1, and characteristics of the recombinant enzyme.CurrMicrobiol39. 351-0357.
- 38.Shi R, Li Z, Ye Q, Xu J, Liu Y. (2013) Heterologous expression and characterization of a novel thermo-halotolerant endoglucanase Cel5H fromDictyoglomusthermophilum.BioresourTechnol142. 338-344.
- 39.J P Euzéby. (1998) Taxonomic note: necessary correction of specific and subspecific epithets according to Rules. 12c and 13b of the International Code of Nomenclature of Bacteria , Revision).Int 48, 1073-1075.
- 40.Brumm P, Hermanson S, Hochstein B, Boyum J, Hermersmann N et al. (2011) MiningDictyoglomusturgidumfor enzymatically active carbohydrases.ApplBiochemBiotechnol163. 205-214.
- 41.P J Brumm, Gowda K, F T Robb, D A Mead. (2016) . The Complete Genome Sequence of HyperthermophileDictyoglomusturgidumDSM 6724 Reveals a Specialized Carbohydrate Fermentor.FrontMicrobiol7,1979 .
- 42.Gao D, Uppugundla N, S P Chundawat, Yu X, Hermanson S et al. (2011) Hemicellulases and auxiliary enzymes for improved conversion of lignocellulosic biomass to monosaccharides.BiotechnolBiofuels4. 5.
- 43.P J Brumm, Hermanson S, Luedtke J, D A Mead, Brumm P et al. (2011) Identification, cloning and characterization ofDictyoglomusturgidumCelA, a thermostable endoglucanase with both cellulase and mannanase activity. .Journalof life sciences. , (Libertyville, Ill.)5 488-496.
- 44.S F Altschul, T L Madden, A, Zhang J, Zhang Z et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs.Nucleic Acids Res25. 3389-3402.
- 45.Notredame C, D G Higgins, Heringa J. (2000) T-Coffee: A novel method for fast and accurate multiple sequence alignment.J. MolBiol302 205-217.
- 46.P Di Tommaso, Moretti S, Xenarios I, Orobitg M, Montanyola A et al. (2011) T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension.Nucleic Acids Res39. 13-17.
- 47.Llados J, Cores F, Guirado F. (2018) . Scalable Consistency in T-Coffee Through Apache Spark and Cassandra Database.JComputBiol25 894-906.
- 48.S F Altschul, T L Madden, A, Zhang J, Zhang Z et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs.Nucleic Acids Res25. 3389-3402.
- 49.Guindon S, Gascuel O. (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood.Systematic biology52. 696-704.
- 50.Dereeper A, Guignon V, Blanc G, Audic S, Buffet S et al. (2008) Phylogeny.fr: robust phylogenetic analysis for the non-specialist.Nucleic Acids Res36. 465-469.
- 51.Chevenet F, Brun C, A L Banuls, Jacq B, Christen R. (2006) TreeDyn: towards dynamic graphics and annotations for analyses of trees.BMC Bioinformatics7. 439.
- 52.Armenteros Almagro, J, K D Tsirigos, C K Sonderby, T N Petersen et al. (2019) SignalP 5.0 improves signal peptide predictions using deep neural networks.Nature biotechnology37. 420-423.
- 53.Spriestersbach A, Kubicek J, Schafer F, Block H, Maertens B. (2015) . Purification of His-Tagged Proteins.Methods in enzymology559 1-15.
- 54.Zhang X, Lei H, Zhu L, Wu J, Chen S. (2015) From lignocellulosic biomass to renewable cycloalkanes for jet fuels.Green Chemistry17. 4736-4747.
Cited by (2)
This article has been cited by 2 scholarly works according to:
Citing Articles:
Enzyme and Microbial Technology (2026) Crossref OpenAlex
Molecules (2021) Crossref Semantic Scholar OpenAlex