
Hepatic Cysts as a Manifestation of Polycystic Kidney Disease (Polycystic Liver Report of 2 Mother-Son Cases)
Abstract
Polycystic kidney disease is an inherited disease that can lead to high blood pressure and kidney failure. In Mexico, 4.5% of patients with kidney failure are carriers of this disease; the liver is another of the organs affected by this disease that can manifest as abdominal pain and a mass effect in the abdominal cavity; we present 2 cases of polycystic kidney and liver disease (mother and child), in addition to describing the clinical manifestations, two different stages of the disease are shown, being a hereditary disease it is suggested that once a case is identified, an abdominal ultrasound is performed to first-degree relatives in search of cystic lesions to indicate preventive measures that help us preserve the overall well-being of the patient.
Article Information
- Received
- Accepted
- Published
Academic Editor: Sasho Stoleski, Institute of Occupational Health of R. Macedonia, WHO CC and Ga2len CC.
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2021 Osnaya-Romero N, et al.
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Corresponding author: Corresponding author: Osnaya-Romero N., Unidad de investigación clínica, Instituto Nacional de Pediatría —
Competing Interests
The authors have declared that no competing interests exist.
Funding
No specific funding statement was provided by the authors.
Data Availability
No data-availability statement was provided by the authors.
Citation:
Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is an inherited disease in which the children of the affected father have a 50% chance of developing the disease; In more than 50% of the patients they present end-stage renal failure, which is the cause of morbidity and mortality in these patients; the prevalence varies from one in 400 to one in 1000, with a dominant inheritance pattern. Within the organs that are affected in this disease in addition to the kidneys is the liver. ADPKD is the most common inherited disease; In the United States, an incidence of 1/1000 inhabitants is reported, in Mexico there is a Méndez-Duran report from 2010 in which it reports that 4.5% of patients with kidney failure have a diagnosis of polycystic kidney disease, however many of these patients develop liver cysts, most of which are not a cause of liver failure 1, 2, 3
When a patient with this diagnosis is identified, first-degree relatives should be told about the risk of developing the disease, and an abdominal ultrasound should be performed in search of kidney and liver cysts or the determination of PKD1 and PKD2 mutations. 2
The following is the case of a woman who is detected polycystic kidney disease, which is why she is transplanted , however, she presents growth of liver cysts during the 18 years after the transplant, cysts that are having a mass effect in this patient, at the same time the case of his son is presented who is diagnosed with polycystic kidney and liver disease
Case 1
A 65-year-old woman from Mexico City with a history of her mother who was diagnosed with polycystic kidney disease at the age of 71, dying 2 years later from complications of the same disease; the diagnosis was corroborated by autopsy study with renal, hepatic and cerebral cysts; a brother of hers died from complications of polycystic kidney disease; a 53-year-old living brother with polycystic kidney disease currently with high blood pressure.
She has 3 sons, 2 healthy sons and the third of her, at age 29, was diagnosed with kidney and liver cysts.
It begins at age 47 with diffuse abdominal pain, due to the mother's history; An abdominal ultrasound (USG) was requested, where kidneys and liver cysts were reported. The findings in the renal function tests showed acute renal failure and peritoneal dialysis was started without success; Subsequently, she underwent a kidney transplant without complications, currently 18 years after the transplant without problems, at age 60 she presented diabetes mellitus; In the last 3 years she started with intermittent abdominal pain, later she noticed abdominal distension, the episodes of pain had increased and on occasions it was accompanied by intolerance to the oral route, she refers to dyspnea on medium exertion; pain treatment was started with non-steroidal analgesics
On physical examination, globose abdomen at the expense of hepatomegaly, collateral venous circulation, painful palpation, the rest of the examination unchanged.
Abdominal tomography reported hepatomegaly secondary to polycystic disease and was reported with 3 cysts of greater size of 17, 14 and 12 cm in approximate diameter (Figure 1, Figure 2), kidney transplant without problems; the liver with mass effect on the stomach, the vena cava partially occluded, uncomplicated colon diverticulosis. Laboratory creatinine 1.44mg/dl urea 51.9 md/dl, glucose 71mg/dl, normal liver function tests.
The current management is metformin 500mg c/ 12 hrs, mycophenolate 500mg c/ 12hrs, losartan 50mgc/ 24hrs, prednisone 5mg c/ 24hrs, omeprazole 20mg c/ 24hrs.
Case 2
33-year-old male, son of the woman from case 1, with diagnosis of polycystic kidney and liver disease (Figure 3, Figure 4) since 29 years old , with normal liver and kidney function; At that moment general measures were started based on a diet low in sodium, increasing the consumption of natural liquids, avoiding nephrotoxic drugs and monitoring blood pressure.
He has presented 1 event of hematuria that spontaneously subsided, an event of pain located in the left flank; 4 years later, he presented arterial hypertension initiating its management with losartan and nifedipine, renal function tests within normal limits.
At physical examination, without alterations. The abdominal thomography reports kidneys in habitual anatomical projection with loss of their morphology and increased sizes with loss of the sinus parenchyma relationship due to cystic-looking structures; right kidney with dimensions 14.3x 8.9 x 6.6 cm left kidney 17.3 x 11.6 x 8.2 cm in its longitudinal, transverse and anteroposterior diameters respectively, poorly determined echogenic renal sinuses, the left renal sinus with solid oval shades measuring 0.6 to 0.8cm on average and some calcifications; liver ultrasound showed cystic lesions. Laboratory creatinine 0.96mg/dl, urea 31.03 mg/dl
Figure 1. Non-contrast CT shows hepatomegaly by multiple clusters of cysts.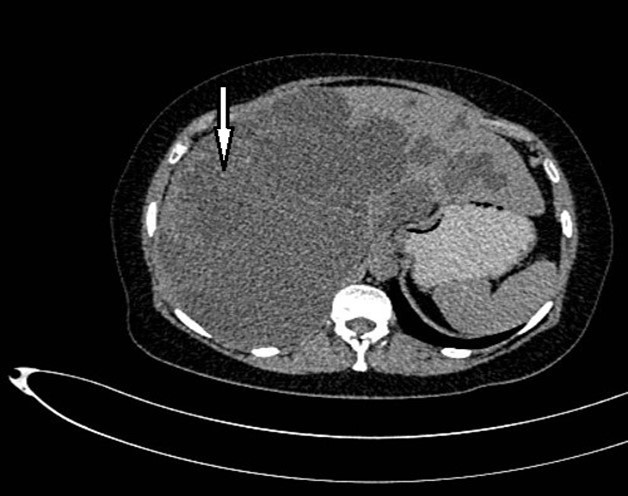
Download figure
Figure 2. Non-contrast CT shows multiple liver cysts with variable size and multiple renal cysts.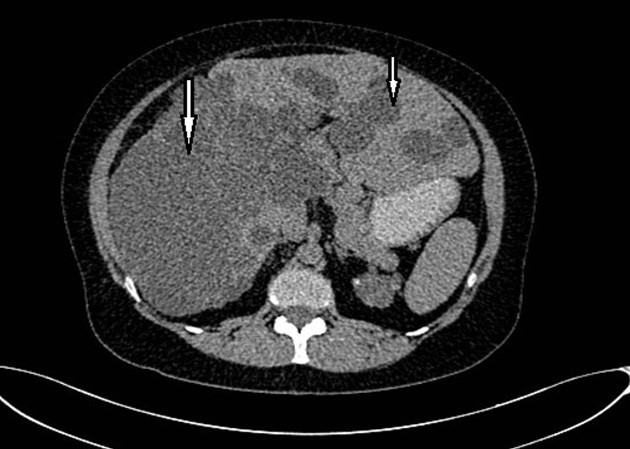
Download figure
Figure 3. Contrast CT coronal reconstruction shows small liver cysts, kidneys are enlarged by multiple cysts.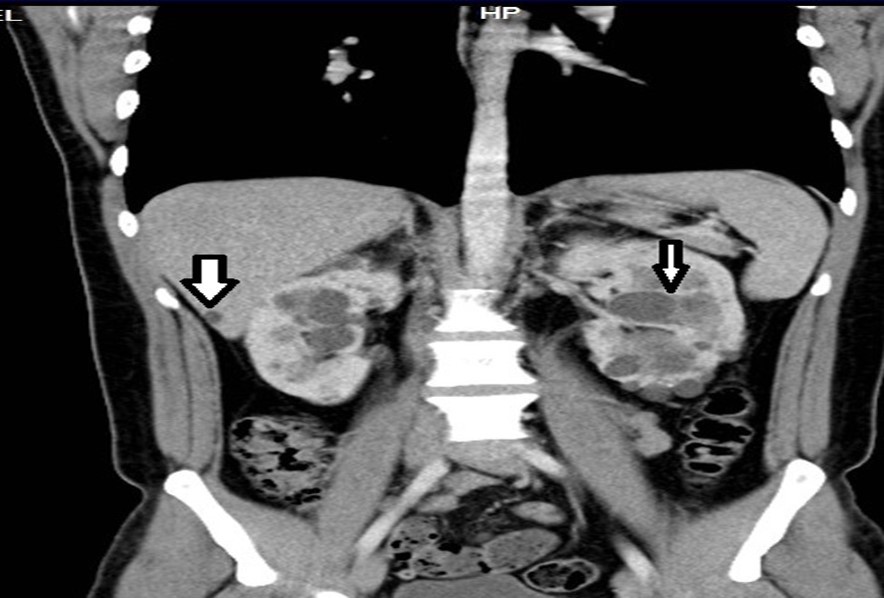
Download figure
Figure 4. Contrast CT shows non complicated small liver cysts and multiple renal cysts.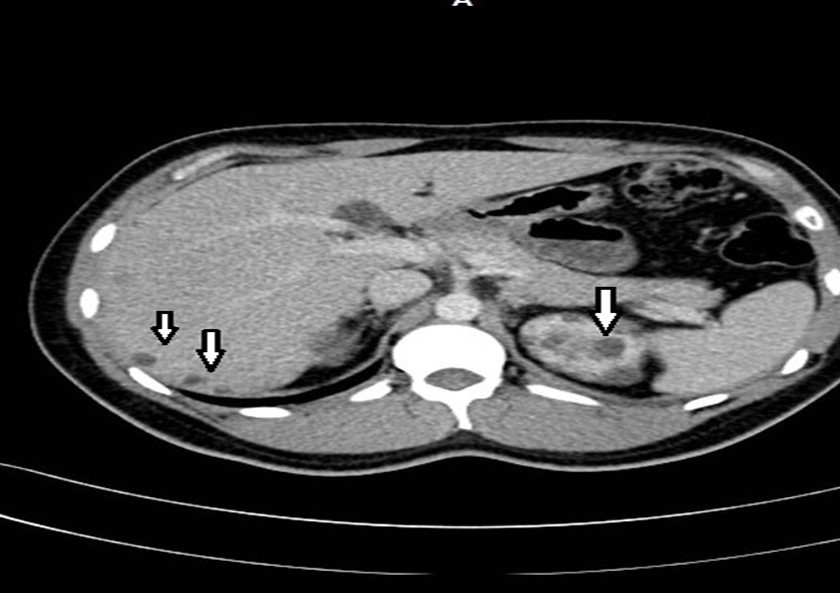
Download figure
Discussion
The diagnosis of ADPKD is suspected in the presence of renal cysts; In these patients, a family history of polycystic disease, arterial hypertension, and lesions in some other organ that may increase morbidity and mortality in these patients, such as cardiovascular and central nervous system lesions, should be sought; In the cases that we present, the diagnosis in the first case was suspected due to the symptoms of abdominal pain and family history; This allowed her to perform a kidney transplant, however, her follow-up did not include the follow-up of liver cysts, which increased in size in such a way that they are the cause of chronic abdominal pain that the patient presents; the second case shows us a diagnosis in the second decade of life, which allows the patient to start with measures for the preservation of kidney function, which is the main organ that affects this disease, being a young patient it is important to carry out surveillance of all the organs in which cysts have been detected, for in order to give a timely management and that the quality of life of these patients is not affected. 4
Diagnosis by ultrasound (USG) of ADPKD type PKD1 is based on the Ravine criteria (In which the presence or not of polycystic kidney disease, the age of the patient and the number of cysts at the time of diagnosis are taken into account), the sensitivity approaches 100% in both patients over 30 years of age and in younger patients. The application of the Ravine criteria in patients with a PKD2 mutation is less, specifically in the group younger than 30 years (in this case, the diagnostic sensitivity is around 67%). In patients with no family history, more precise critical diagnostic criteria developed by Demetriou et al. (Take into account the age of the patient and the number of cysts identified by renal USG) and are recommended in suspected cases of PKD2-type ADPKD. 5, 6
Pei et al. they proposed a classification for the ultrasonography identification of both genotypes and for the genotype unknown; this rating offers positive predictive value as well as sensitivity. 7
Cystic lesions form in both kidneys causing progressive deterioration in kidney function, and there may be cystic lesions in other organs, such as the liver (30-94%), seminal vesicles (40%), brain aneurysms (18-40%)) diverticular disease (20-25%), mitral valve prolapse (25%), pancreas (10%), spleen (5%), arachnoid space (8%) other less frequent lesions are cysts in the thyroid, ovary, endometrium, lung, pituitary gland, breast, epididymis, aneurysm of the abdominal aorta, wall hernias and hypertrophy of the left ventricle. The vascular lesions such as intracranial aneurysms, dolichoectasias, dissections of the aorta and other arteries are injuries that represent an increased risk of morbidity and mortality in these patients. 8, 9, 10, 11.
The clinical manifestations of polycystic liver disease are generally due to the mass effect, causing abdominal pain, gastroesophageal reflux, vomiting and sometimes dyspnea, among other symptoms; These symptoms are only observed in patients in whom the cysts acquire a considerable size in such a way that they function as an intra-abdominal mass; In most patients with polycystic kidney disease, hypertension and hematuria are the first symptoms that increase the risk of kidney damage; Renal failure occurs in the fourth and sixth decade of life, the cysts that grow more frequently are renal cysts, liver cysts are generally asymptomatic, so the follow-up of these patients is generally in charge of the nephrologist, the cause of pain in these patients is located in the iliac fossa or in the two-lumbar region that may be associated with urinary tract infection or the presence of kidney stones, in this patient, the cause of abdominal pain is given by the compression exerted by the liver growth due to the presence of cysts compressing other intra-abdominal organs; In the patient, the problem of kidney failure occurs in the fourth decade of life, a kidney transplant was performed, however the growth of liver cysts continues during these years causing an increase in abdominal volume and an intra-abdominal mass effect which are the origin of chronic abdominal pain that the patient has presented for 3 years, pain that has been increasing and is sometimes disabling, the initial management with non-steroidal anti-inflammatory drugs and currently pain management is based on tramadol.. 1, 13, 14
The diagnosis in these patients is with a hepatic and renal USG, the genetic diagnosis is not mandatory in all cases, but in relatives who may be potential donors it is necessary to carry out the genetic diagnosis 8.
So far, 2 mutations have been described in patients with PKD1 and PKD2 polycystic kidney disease. Both mutations produce renal and extrarenal manifestations, however both cases develop kidney failure at different age; in PKD1 gene kidney failure appears at an average age of 54.3 years, and in more than 50% of patients with PKD 2 develop kidney failure later. 15, 16, 17, 18
The presentation of these two cases allows us to illustrate the evolution of ADPKD in 2 different stages, in case 1 the diagnosis is suspected due to the symptoms and the history of the mother who died of ADPKD at the time of diagnosis he had kidney failure, so after trying unsuccessful peritoneal dialysis, he underwent a kidney transplant with a good quality of life, however the evolution of the disease has continued in other organs, so at this time he has secondary hepatomegaly to hepatic cysts that is conditioning a mass effect on the intra-abdominal organs causing abdominal pain, postprandial vomiting; the elevation of the right hemidiaphragm, secondary to hepatomegaly, could explain the dyspnea on medium efforts; pain is disabling that has been managed with tramadol.
The literature mentions that liver cysts are usually asymptomatic in up to 80% of cases, however in case 1 the liver cysts have increased in size causing abdominal pain, the increase in cysts in women may be related to the stimulus exerted due to estrogens.
The treatment suggested by some authors is partial liver resection, fenestration, aspiration, sclerotherapy, or liver transplantation, which may be a therapeutic option in some cases; In this patient, it has not been possible to perform any of these treatments due to poor control of diabetes and her glomerular filtration rate is less than 50%, which is why surgery is awaited 19, 20, 21.
Currently the patient is in good condition, waiting to be evaluated by surgery to offer her an option regarding liver cysts.
In case 2, the patient is asymptomatic so far in relation to liver cysts; has presented arterial hypertension, so management has focused on this problem, it is thought that there are 2 hypotheses about the presence of arterial hypertension, one is that there is a malfunction of the endothelium and renal vessels, activating the renin angiotensin system, which results in arterial hypertension up to 60% and even maintaining normal renal function, the second hypothesis about the pathogenesis of arterial hypertension is the decrease in polycystin 1 or 2 in the cilia of endothelial cells and in muscle smooth vessels, decreasing Ca2 + -dependent nitric oxide levels, which can lead to an increase in blood pressure.
Taking into account the evolution of the first case and that it is a young patient, the first problem to be solved is the preservation of kidney function while monitoring the growth of liver cysts in such a way that early treatment is carried out to avoid its growth and that represents a problem for the quality of life of the patient2, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22 .
Based on the above, the management of arterial hypertension in this patient is with renin angiotensin system blockers, renal function should be monitored; In the case of calcium blockers, some studies reported a decrease in glomerular filtration rate, so it should not be used first, however, they are used in resistant hypertension; the use of diuretics is controversial due to their action on the renin angiotensin system, some authors suggest that they should not be first line.22
The follow-up of these patients should be carried out with creatinine clearance, renal and hepatic USG, in the first case, it will serve to monitor the function of the kidney transplant and the growth of hepatic cysts; and in case 2 it should be done to monitor kidney function and perform a kidney transplant in a timely manner; Ultrasound monitoring allows us to monitor cystic lesions in both the kidney and the liver, which, if not received timely treatment, can affect the quality of life of the patient even when a kidney transplant can be performed. 8
Conclusion
Polycystic kidney disease is one of the main inherited kidney diseases that causes kidney failure; when a patient with polycystic kidney disease is identified, the family should be studied to look for new cases; Ultrasound is helpful in monitoring progress and looking for the presence of cysts in other organs such as the liver and pancreas and making treatment decisions before complications occur. In the case of having relatives of pediatric age, knowing that 50% of each child may have the disease, the search for the PKD1 and PKD2 gene can be an alternative for early diagnosis; however, this technique is not available in all hospitals and this limits its use for diagnosis.
Some authors comment that it is unlikely to find cysts in patients under 30 years of age, in case 1 she was present renal failure in the fourth decade of life, which implies that the onset with alterations between the second and third decade of life; The transplant solved the problem of kidney failure, however the liver cysts grew to exert intra-abdominal pressure causing episodes of chronic abdominal pain that until now has only been managed with analgesics without being able to offer another management option; case 2 is diagnosed in the second decade of life with complications such as hypertension, which implies that cystic lesions begin at an early age, these 2 cases are an example of polycystic disease that begins between the second and third decades of life, causing high blood pressure as the first manifestation of kidney damage to kidney failure in the fourth decade of life, however the growth of liver cysts although they have not caused alterations in liver function, if they are the cause of chronic abdominal pain and effect of intra-abdominal mass for which these patients should have a comprehensive follow-up
Therefore, as soon as a case is diagnosed, first-line family members should be screened for early cases. Having an early diagnosis helps preserve kidney function, however, polycystic disease is a progressive disease and if there are cysts in other organs, these lesions can also cause other symptoms depending on the affected organ, so the monitoring of these patients should be comprehensive
References
- 1.Panozo Borda SV, Heredia Moy K, Oviedo Gamboa I, Villarroel Arze T, Zegara Santiesteban W et al. (2012) Estudio imagenologico de poliquistosis renal autosómica dominante, reporte de un caso y revisión de la literatura.Gac.Med. , Bol 35(1), 31-34.
- 2.Ars E, Bernis C, Fraga G, Martinez V, Martins J et al. (2015) Guías Clínicas Españolas de Poliquistosis renal Autosómica Dominante. https://escolasaude.sergas.es/Docs/EGSPC/pilula/Poliquistosis/resources/doc02.pdf
- 3.Méndez-Durán A, J F Méndez-Bueno, Tapia-Yáñez T, Muñoz Montes A, Aguilar-Sánchez L. (2010) Epidemiología de la insuficiencia renal crónica en México.Dial-Trasp. 31(1), 7-11.
- 5.Gradzik M, Niemczyk M, Golebiowski M, Paczek L. (2016) Diagnostic Imaging of Autosomal Dominant Polycystic Kidney Disease. Pol. 81, 441-53.
- 6.Ravine D, Gibson R N, Walker R G, Sheffield L J, Kincaid-Smith P et al. (1994) Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. , Lancet 343, 824-7.
- 7.Pei Y, Obaji J, Dupuis A, Paterson A D, Magistroni R et al.Unified criteria for ultrasonographic diagnosis of ADPKD. , Journal of the American Society of Nephrology. JASN 20(1), 205-12.
- 8.O Tobal D Noboa. (2014) Poliquistosis renal autosómica dominante. Necesidad de diagnóstico y tratamiento oportuno. Rev med Urug. 30(3), 184-192.
- 9.Pirson Y, Chauveau D, Torres V. (2002) Management of cerebral aneurysms in autosomal dominant polycystic kidney disease. , J Am Soc Nephrol 13(1), 269-76.
- 10.M J Torres, C Rodríguez Pérez J, R Hernández Socorro C, Anabitarte Caballero A, Vázquez C et al. (2006) Diagnóstico molecular de la poliquistosis renal autosómica dominante en la comunidad autónoma de canarias. , Nefrología 26(1), 666-672.
- 11.Akoh J A. (2015) Current management of autosomal dominant polycystic kidney disease. , World J Nephrol 4(4), 468-79.
- 12.Igarashi P, Somlo S. (2002) Genetics and pathogenesis of polycystic kidney disease. , Journal of the American Society of Nephrology 13(9), 2384-98.
- 13.Torres V E, Harris P C, Pirson Y. (2007) Autosomal dominant polycystic kidney disease. The Lancet. 369, 1287-381.
- 14.Martínez Morcillo A, MA Esteban de la Rosa, P De Diego Fernández, García González M, Fernández Castillo M. (2018) Argüelles Toledo I et al. Panorámica de la poliquistosis renal autosómica dominante en una región del sur de España. Nefro. 38(2), 190-196.
- 15.Hateboer N, Dijk M A, Bogdanova N, Coto E, Saggar-Malik A K et al.Comparison of phenotypes of polycystic kidney disease types 1 and 2. European PKD1-PKD2 Study Group. Lancet 353(9147), 103-107.
- 16.Fraile Gómez P, Corral Moro E, García-Cosmes P, Gonzalez Sarmiento R.Tabernero Romo JM. Genetic analysis (PKD2) of autosomal dominant poliycystic kidney disease. , Nefrología 29(6), 562-568.
- 17.García-Garces M, Sánchez Zavala J, Mckinney Novelo I, Corrales Rosas B, Zavala García C et al. (2011) Enfermedad poliquística hepática asociada a enfermedad poliqística renal autosómica dominante. Rev Invest Med Sur. 18(3), 132-135.
- 18.M V Irazabal, V E Torres.Poliquistosis renal autosómica dominante .Nefrología suplemento extraordinario. 2011; 2(1) :. 1-33.
- 19.Martinez-Perez Alberola Soler A, C Domingo del Pozo, Permartin-Comella B, Martinez -Lopez E, Vazquez Tarragon A. (2016) Laparoscopic surgery and polycistic liver disease: Clinicopathological features and new trend in management. , J Min Access Surg 12, 265-70.
- 20.van Aerts RMM, LFM van de Laarschot, Banales J M, JPH Drenth. (2018) Clinical management of polycystic liver disease. J Hepatol. 68(4), 827-837.