
Resolution of Fetal Hydrops in a Case of Congenital Pulmonary Airway Malformation
Abstract
Congenital pulmonary airway malformation (CPAM) can often be noted on fetal ultrasound. When a CPAM is noted in the face of fetal hydrops, the outcome is dismal. Surgical interventions and administration of antenatal steroids can be considered for treatment, but in hydropic fetuses these interventions are not always successful. We present a case of CPAM with severe fetal hydrops that underwent steroid administration and multiple intrauterine surgical interventions, which resulted in an excellent neonatal outcome.
Author Contributions
Academic Editor: Ramesh Bhat Y, Department of Pediatrics, Kasturba Medical College, Manipal University, Manipal, Udupi District, Karnataka, India.
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2018 Alexandria J. Hill, et al
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Introduction
Congenital pulmonary airway malformation (CPAM), previously called congenital cystic adenomatoid malformations (CCAM), is a rare heterogeneous type of lung lesion with an unknown etiology that arises from anomalous growth of the lung buds 1, 2, 3. These lesions consist of cystic or non-cystic lesions of uniform or varying sizes that were previously identified as one of three groups based primarily on the size of the cysts 4, Table 1. Currently using the size of the mass, as well as gross and histologic appearance, a CPAM can be classified as type 0 through 4, listed in Table 21, 2, 3. The incidence of CPAM is approximately 1:11,000 to 1:35,000 pregnancies 2, 3and accounts for approximately 95% of all congenital cystic lung disease 1.
Table 1. Original CPAM Classifications| Type | Cyst Description | Histologic Findings |
| I | -Single or multiple | - Cysts lined by ciliated pseudostratified columnar epithelium |
| - Large | - Walls with smooth muscle and elastic tissue | |
| - >2cm in diameter | -Mucus present in 1/3 of cases | |
| II | - Multiple | - Cysts lined by ciliated cuboidal to columnar epithelium |
| - Small | - Structures resemble respiratory bronchioles and distended alveoli present | |
| - <1cm in diameter | ||
| III | - Large | - Non-cystic lesion producing mediastinal shift |
| - Bulky | - Bronchiole-like structures lined by ciliated cuboidal epithelium and separated by masses of alveolus-sized structures lined by non-ciliated cuboidal epithelium |
| Type | Gross Appearance | Microscopic appearance | Frequency | Presumed site of development | Cystsize |
| 0 | Solid. Lungs are small and firm throughout | Ciliated pseudostratified cyst lining the bronchial–like structures with connective cartilage, smooth muscle and glands | 1-3% | Tracheobronchial | 0.5 cm |
| 1 | One or more large cysts | Cysts lined with cuboidal to pseudostratified columnar cells; large cysts with smaller cysts with walls of fibromuscular tissue, and cartilaginous plates/islands | 50-60% | Bronchial or bronchiolar | 2-10 cm |
| 2 | Sponge-like multiple small cysts with solid pale tumor-like tissue | Cyst lining of cuboidal to columnar cells, ciliated, resembling ectaticbronchiole-like structures, separated by normal alveoli; striated muscle in 5% | 20-25% | Bronchiolar | <2-2.5 cm |
| 3 | Solid | Excess of bronchiolar structures with cyst lining of ciliated cuboidal cells resembling fetal lung in canalicular stage | 8% | Bronchiolar/ alveolar | <0.2 cm |
| 4 | Large cysts in peripheral lung | Cysts lined by flattened epithelium resting on loose mesenchymal tissue, resembling bullous emphysema | 10% | Distal acinar | Up to 10cm |
The clinical presentation of CPAM is variable, and primarily detected by antenatal ultrasound 1.Mediastinal shifts, polyhydramnios, hydrops, or intrauterine death have been seen in large CPAM lesions. Smaller lesions can go undetected with no signs of complications or respiratory symptoms at birth 1. There is a 0.7% risk of malignant transformation within the cyst and impending demise in over 85% of cases that present with fetal hydrops 1, thus early diagnosis is crucial. Prenatal interventions can include thoraco-amniotic shunts, fetal surgery, maternal steroids 1, and ex utero intrapartum treatment (EXIT) procedures.Antenatal monitoring can be achieved by following the CPAM volume to head circumference ratio (CVR), calculated by (length x height x width of the mass x 0.52) divided by the head circumference. This measurement allows for a gestational age-corrected volume ratio. A CVR at or above 1.6 is predictive of an 80% risk of hydrops, whereas a CVR less than 1.6 carries a mere 2% risk of hydrops 5. Symptomatic neonates require surgery, whereas the treatment for asymptomatic neonates still remains controversial 1.
Case
A 33-year-old gravida 1 initially presented for a detailed ultrasound in our office with fulminant fetal hydrops and a noted mass in the left chest. Initial evaluation included maternal viral studies (negative parvovirus, CMV, toxoplasmosis), maternal karyotype (46XX), and amniocentesis (46XX, microduplication in the 16p11.2 region). Given concern for CPAM and hydrops at 20 weeks and 3 days, the patient was referred to a fetal therapy center. Ultrasound at the fetal therapy center showed an estimated fetal weight of 824 grams (>97th percentile) and fetal hydrops (scalp edema of 8mm, skin edema of 4mm, and ascites of 14mm). The diaphragm was intact and the maximal vertical pocket was 6.8 cm. The left chest mass measured 6.6 x 5.3 x 4.4 cm and was multiloculated with microcystic and macrocystic lesions (type 1); the dominant cyst measured 4.0 x 3.2 cm, Figure 1. Secondary to mass effect, the heart was severely displaced into the right chest with a CVR of 3.9cm2. There was no noticeable peristalsis of the cystic structures and no systemic identifiable arterial blood supply. Fetal echocardiogram showed a structurally normal heart significantly shifted in the right chest. After extensive counseling, the patient opted for administration of antenatal corticosteroids and thoraco-amniotic shunt placement. The patient received betamethasone 12mg intramuscularly prior to shunt placement at 21 weeks and 4 days and 21weeks 5 days. Two double pigtail Harrison thoraco-amniotic shunts were placed by inserting a 13 Ga cannular-trocar into the maternal abdomen and uterus (the first shunt into the largest cyst (left, anterior, inferior) and the second into an additional dominant cyst (anterior, central portion of the mass). Both cysts drained completely after insertion. Epidural anesthesia with IV sedation were used as well as tocolytics as needed during the procedure. The post-procedure CVR was 1.3cm2 with decreased mass effect on the heart. Post-operative plan consisted of genetic consultation, fetal MRI, and serial monitoring of the CVR.
Figure 1.CPAM noted at initial fetal surgery consultation; sagittal view on the left and transverse view on the right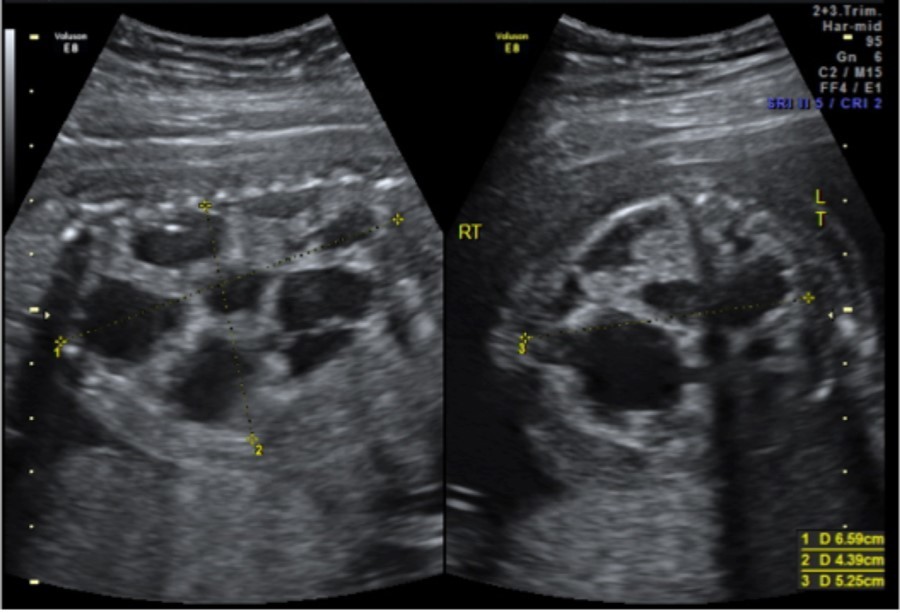
Follow-up parental microarray showed that the 16p11.2 microduplication was maternally inherited. This duplication can be linked with an increased risk of autism or other early onset psychological or neurodevelopmental abnormalities. Fetal MRI was performed at 25 weeks 5 days, noting a large left hemithorax multicystic lesion consistent with CPAM as well as right lung hypoplasia, fetal hydrops, and polyhydramnios. Serial ultrasounds following shunt placement (Figure 2) and CVR (Table 3) noted dislodged shunts on three occasions. With the dislodged shunts, the dominant cyst significantly increased, as did the CVR. On these three occasions (at 24 weeks 3 days, 28 weeks 3 days, and 31 weeks 3 days) repeat thoraco-amniotic shunts, two per procedure, were placed. Immediately after each shunt placement the CVR was notably decreased (Table 3). Amniodrainage was performed at shunt placement when necessary.
Figure 2.Two thoraco-amniotic shunts can be seen in the dominant CPAM cyst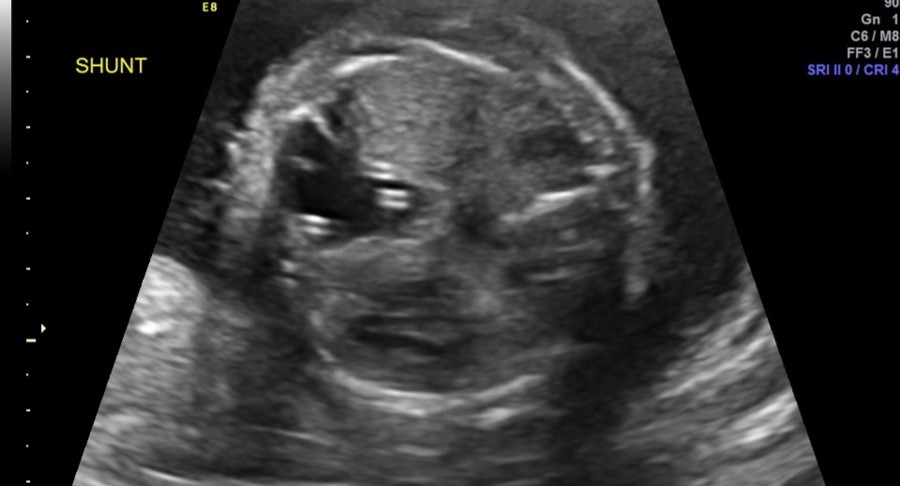
| Gestational Age | CVR (cm2) | Shunts |
| 22w1d | 3.9 | 2 placed |
| 22w2d | 1.3 | In place |
| 23w2d | 2.9 | Both displaced |
| 24w1d | 3.1 | 2 placed |
| 24w3d | 1.0 | In place |
| 25w3d | 1.1 | In place |
| 26w3d | 0.9 | In place |
| 27w5d | 2.0 | Both displaced |
| 28w1d | 2.9 | 2 placed |
| 28w3d | 1.2 | In place |
| 29w2d | 0.9 | In place |
| 30w6d | 2.4 | Both displaced |
| 31w2d | 3.1 | 2 placed |
| 31w4d | 1.2 | In place |
| 32w5d | 1.3 | In place |
| 33w3d | 1.6 | In place |
| 34w5d | 1.2 | In place |
| 35w5d | 0.74 | In place |
| 36w3d | 1.35 | 1 in place |
On ultrasound at 36 weeks 6 days two shunts were clearly identified in the fetal chest, the amniotic fluid level was 28cm, and the CVR was 1.5cm2. Six shunts were seen free floating in the amniotic cavity. The dominant cyst measured 2.5 x 1.3 x 0.9 cm with no evidence of fetal hydrops. Regular antenatal testing was reassuring until delivery, which was performed via cesarean at 37 weeks and 2 days. At delivery one thoracic shunt was noted to be intact in the fetal chest.
The infant, born weighing 3,015 grams, was intubated at 1 minute of life due to noted bradycardia and no respiratory effort. With positive pressure ventilation on 100% oxygen, the infant had oxygen saturations of 80% and a heart rate of 150. Apgar scores were 6 and 7 at 1 and 5 minutes respectively. The neonate ultimately required vasopressor support and a left sided tension pneumothorax was noted requiring chest tube placement. Thoracotomy, Figure 3, was performed at 21 hours of life to remove the mass. On post-operative day one the neonate had improved hemodynamics and decreased ventilation pressures with ongoing vasopressor support. The neonate was extubated on day of life 19 and ultimately discharged from the NICU on day of life 34, weighing 3.87 kilograms, with oxygen (1/8 L at 100%) via nasal cannula, continued for 56 days. She was meeting appropriate milestones and scoring well on her Ages and Stages Questionnaires at 4 and 6 months. At 7 months of age she weighed 17lb 15oz and was meeting all appropriate milestones for her age.
Figure 3.Neonatal thoracotomy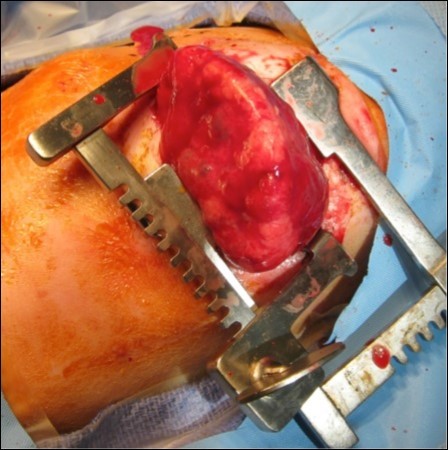
Comment
Fetuses diagnosed with hydrops secondary to CPAM will likely die if no intervention is made. Retrospective studies have shown that death is the ultimate outcome of all of fetuses that presented with hydrops and underwent expectant management alone 6. When thoraco-amniotic shunts are the method of treatment, data has shown that one procedure often decompresses the cystic lesion 7. This retrospective review discussed 11 CPAM cases, 6 with hydrops, which had thoraco-amniotic shunts placed. All were successful after the first attempt in decompressing the dominant cyst and ultimately resulted in vaginal delivery (with a mean gestational age of 38.2; range 35.6 – 40.0) with the exception of one fetus that demised one day after shunt placement. Of note, this fetus had a CVR of 3.2cm2 at 17 weeks (for reference, our patient presented with hydrops and a CVR prior to first shunt placement of 3.9 cm2). All had thoracotomy after delivery, and all but one noted the shunt to be in place. Also, administration of antenatal corticosteroids has been shown, particularly in those with microcystic CPAMs, to reduce the CVR and possibly resolve fetal hydrops 8. Given that the lesion in our case had microcystic components with early hydrops, steroids were administered. Our steroid administration was earlier than the gestational age of those in the study given the severity of the hydrops at such an early gestational age.
In the literature, multiple thoraco-amniotic shunt placements to treat CPAM were not found. In CPAM cases presenting with advanced hydrops, aggressive surgical treatment with thoraco-amniotic shunts is reasonable with appropriate preoperative workup and patient counseling. The typical expectation can be resolution of the dominant cyst after one procedure. This case illustrates that multiple procedures may indeed be necessary and can be successful in maintaining a stable dominant cyst, a low CVR, and resolution of hydrops. Although unconventional, these multiple procedures allowed an initially hydropic fetus to be born, at a term gestation, without any signs of hydrops and undergo thoracotomy after delivery with no complications.
References
- 1.Pelizzo G, Costanzo F, Andreatta E, Calcaterra V.Congenital pulmonary airway malformations: from prenatal diagnosis to postnatal outcome. Pediatrician update. Minerva Pediatr.2015Sep11.[Epub ahead of print] PubMed PMID:. 26365821.
- 2.Bolde S, Pudale S, Pandit G, Ruikar K, Ingle S B.Congenital pulmonary airway malformation: A report of two cases. World J Clin Cases.2015May16; doi: 10.12998/wjcc.v3.i5.470. PubMed PMID: 25984523; PubMed Central PMCID: PMC4419112 3(5), 470-3.
- 3.Baral D, Adhikari B, Zaccarini D, Dongol R M, Sah B.Congenital Pulmonary Airway Malformation in an Adult Male: A Case Report with Literature Review. Case Rep. Pulmonol.2015;2015: 743452. doi: 10.1155/2015/743452. Epub2015Jul8. PubMed PMID: 26236529; PubMed Central PMCID: PMC4510255 .
- 4.Stocker T J, Manewell J E, Drake R M. (1977) Congenital cystic adenomatoid malformation of the lung: classification and morphologic spectrum. , Hum Pathol 8, 155-71.
- 5.Stoiber B, Moehrlen U, Kurmanavicius J, Meuli M, Haslinger C et al.Congenital Lung Lesion: Prenatal Course, Therapy and Predictors of Perinatal Outcome. Ultraschall Med.2015Jun30. [Epub ahead of print] PubMed PMID:. 26126151.
- 6.Adzick N S, Harrison M R, Crombleholme T M, Flake A W, Howell L J. (1988) Fetal lung lesions: management and outcome. , Am J Obstet Gynecol 179, 884-889.
- 7.Schrey S, Kelly E N, Langer J C, Davies G A, Windrim R et al. (2012) Fetal thoracoamniotic shunting for large macrocystic congenital cystic adenomatoid malformations of the lung. Ultrasound Obstet Gynecol Published online in Wiley Online Library (wileyonlinelibrary.com). DOI: 10.1002/uog.11084 39, 515-520.