
Abstract
Trichoderma reeseiβ-glucosidase (Bgl1) is one of four enzymes demonstrated to act synergistically to degrade cellulose both in vitro and in vivo. Our work attempted to better understand the substrate specificity and potential biotechnological applications of Bgl1. T. reesei Bgl1H cleaves over 80% of the β-(1-4) and β-(1-3) linkages in β-glucan and 14% of the β-(1-4) linkages in amorphous cellulose, significantly more than any tested bacterial β-glucosidase. Bgl1H cleaves 50% of the β-(1-4) linkages in xyloglucan when supplemented with cellulase and α-xyloside. Approximately 20% conversion to glucose was obtained from insoluble β-(1,3)-linked curdlan using only Bgl1H; addition of a curdlanase resulted in conversion of approximately 70% of the curdlan to glucose. Bgl1H also produces xylose from xylooligosaccharides and debranched xylans. For both glucans and xylans, the relative rates of hydrolysis increase with increasing polysaccharide chain lengths. Bgl1H is able to partially degrade β-glucan in a variety of grain components; addition of endo-acting enzymes improved the enzyme’s performance on these grain components. The ability of this enzyme to produce monosaccharides from undigestible polysaccharides suggest it may have potential in improving utilization of carbohydrates in animal feed, fermentations, and other biotechnological applications.
Author Contributions
Academic Editor: Mezni Ali, Department of Life Sciences, University of Carthag, Tunisia
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2019 Phillip Brumm, et al.
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
PB is founder and CEO of C5-6 Technologies LLC, a company started to make the biomass-degrading enzymes developed by the GLBRC available to researchers at a nominal charge. The company was formed after the completion of the work presented here, and was not involved in the study design, collection, analysis and interpretation of data.
Citation:
Introduction
Plant-based biomass is made up of three main components, cellulose, hemicelluloses, and lignin. Conversion of this biomass to fuels and chemicals requires the degradation of the cellulose and hemicelluloses to monomer s via enzymatic, chemical, or thermal processes. A number of enzymatic systems have been described for achieving this conversion, including systems from bacterial 1 and fungal 2, 3, 4, 5 sources. The Trichoderma system for cellulose degradation was originally thought to involve a core set of four enzymes: cellobiohydrolase I (CBHI) 6, cellobiohydrolase II (CBHII) 7, endoglucanase1 (Eg1) 8, and β-glucosidase A (Bgl1) 9, 10. An additional type of enzyme, lytic polysaccharide mono-oxygenases are now believed to assist hydrolysis by oxidatively breaking linkages in crystalline regions 11, 12, 13, 14
Bgl1 is reported to cleave (1,2)-, (1,3)-, (1,4)-, and (1,6)-β-D-linked disaccharides displaying the highest specificity for (1,3)-β-D-linked laminaribiose 15. The crystal structure of the enzyme was determined 15 and submitted to the PDB (DOI:10.2210/pdb4i8d/pdb). Researchers have described Bgl1 as the bottleneck in cellulose degradation by the Trichoderma system, and a number of methods have been evaluated for eliminating this bottleneck including hyperexpression of the enzyme 16, expression of fusion of an exotic β-glucosidase with CbhI 17, and hyperexpression of an exotic thermostable β-glucosidase in the organism 18. This enzyme
As part of a project for the Great Lakes Bioenergy Research Center (GLBRC) we produced large quantities of the Trichoderma cellulose-degrading and xylan-degrading enzymes in Pichia pastoris for evaluation using GenPlat 19. Due to the low productivity of the original Bgl1 clone, we had codon-optimized versions of the Bgl1 and a CBM2-Bgl1 fusion protein synthesized and cloned into Pichia pastoris. These two proteins were produced, purified, and compared to each other and bacterial beta-glucosidases for specificity and activity. These comparisons revealed properties significantly different from other beta-glucosidases. The physiological substrates for Trichoderma β-glucosidase is probably not cellobiose, but rather partially hydrolyzed hemicelluloses including β-glucans and xylans.
Material and Methods
Materials
Galactan, galactomannan, arabinoxylan, arabinan, β-glucan, and D-Xylose and D-Glucose (GOPOD Format) Assay Kits were obtained from Megazyme International (Wicklow, Ireland). 2-Nitrophenyl-β-D-glucopyranoside, 4-methylumbelliferyl-β-D-glucopyranoside (MUG), 4-methylumbelliferyl-β-D-cellobioside (MUC), 4-methylumbelliferyl-β-D-manopyranoside (MUM), 4-methylumbelliferyl-β-D-galactopyranoside (MUGal) and 4-methylumbelliferyl-β-D-xylopyranoside (MUX) were obtained from Research Products International Corp (Mt. Prospect, IL). 20. Geneticin (G418), 4-nitrophenyl-β-D-galactopyranoside, 4-nitrophenyl-β-D-mannopyranoside, Avicel PH-101, Whatman 1 filter paper, glucose, and xylose were purchased from Sigma-Aldrich (St. Louis, MO). Xylooligosaccharides were obtained from Cascade Analytical Reagents and Biochemicals (Corvallis, Oregon). Xylanase 4 (Cthe_2972), α-arabinofuranoside 1 (G11MC16_1557), β-glucosidase 1 (Aaz_81839), β-glucosidase 2 (Cthe_0212, Cthe BglA),
β-glucosidase 4 (Bcel_0282), β-glucosidase 5 (Bcel_0705), cellulase 4, (Cthe_0797), curdlanase 1 (Bcell_0683),β-xylosidase 1 (Y412MC61_2711), A. niger α-xylosidase 21, xyloglucanase 2, Sfla_0771, 0797are products of C5-6 Technologies LLC (Fitchburg, WI). Barley flour, oatmeal and oat bran, wheat flour and wheat bran (Bob’s Red Mill, Milwaukie, OR) were obtained from local grocery stores. Phosphoric acid swollen cellulose (PASC) was prepared from Avicel PH-101 by completely dissolving the cellulose in concentrated phosphoric acid 20, 22, followed by precipitation into 10 volumes of deionized water. All other chemicals were analytical grade and used without any further purification.
Glucose production was measured using the Megazyme GOPOD glucose kit and xylose production was measured using the Megazyme K-XYLOSE xylose kit, both using the manufacturer’s instructions. For rate determinations, monosaccharide release was determined using 200 ml of either 1 mM solutions of oligosaccharides or 1% solutions of saccharides in 100 mM acetate buffer, pH 5.0 and 50°C. Substrates were incubated with 85 micrograms of pure enzyme for 10 minutes, the reaction was stopped by incubation at 95°C, and monosaccharides were measured using the appropriate Megazyme kit. For xylooligosaccharide hydrolysis experiments, monosaccharide release was determined using 1000 ml of 1% in 100 mM acetate buffer, pH 5.8 and 50°C. For polysaccharide hydrolysis experiments, monosaccharide release was determined using 1000 ml of 0.2% polysaccharide in 100 mM acetate buffer, pH 5.8 and 50°C. For grain and grain fractions, 10 g samples of material were incubated in 90 ml of 50mM acetate buffer, pH 5.8, at 50°C and 60 rpm in 250 ml bottles. In addition, all long-term reactions contained 5mg/ml vancomycin and 10mg/ml carbenicillin to prevent microbial growth. Enzyme dosing unless noted elsewhere was 20 mg/ml of pure enzyme. Aliquots of the reaction mixture were removed, the reaction was stopped by incubation at 95°C, and monosaccharides were measured using the appropriate Megazyme kit.
Protein purity was determined by SDS PAGE. The specific activity of the purified enzymes was determined using 2-NP-β-D-glucopyranoside at pH 5.0 and 50°C 23; one β-glucosidase unit produces 1 micromole of o-nitrophenol per minute at 50°C from a 1mM solution of 2-nitrophenyl-β-D-glucopyranoside. Protein concentration were determined by A280 spectrophotometry. The extinction coefficient for the enzymes was calculated using ProtParam (https://web.expasy.org/protparam/ ).
Results
Enzyme Cloning and Production
DNA2.0 (now Atum) optimized the CBM1-Bgluc ORF for expression in Pichia pastoris and synthesized the gene based on the sequence determined in previous work 24. The optimized ORF for the CBM1-Bgluc was sub-cloned into BioGrammatics pJANaMF (AOX1 promoter, Nat resistance, alpha Mating Factor, aMF) as a single fragment with BsaIsites.
The 6His-Bgluc was sub-cloned into pJAGaMF (AOX1 promoter, G418 resistance, aMF) as two fragments. The 3’ fragment came from the DNA2.0 clone and extends from a native NcoI site 21 bases 3’ from the end of the CBM1 coding sequence, to the BsaI site at the stop codon. The 6His-Bgluc 5’ fragment was created by annealing the following primers (lower case nucleotides remain single stranded for subcloning):
Bgluc5frag-top:5-ggctCACCACCATCACCACCATGTCGTTCCTCCAGCTGGTACTC-3’
Bgluc5frag-bot:5’-catgGAGTACCAGCTGGAGGAACGACATGGTGGTGATGGTGGTG-3’
The entire N-term fusion and Bgluc coding region and cloning junctions of both expression vectors were sequenced and found to be as predicted for both expression vectors.
Sequencing primers:
1) Bgluc-1860F: 5’- ACACTATTGCTAAGTCGCC-3’
2) Bgluc-1240F: 5’- TGCTAGAGATGGTATTGTG-3’
3) Bgluc-660F: 5’- GCGTTGACCCTTATCTGA-3’
4) Bgluc-1540R: 5’-CAACATCTTTACCTCTAGCG-3’
5) Bgluc-920R: 5’- CCCAACTGATCCTTCAACAC-3’
6) Bgluc-360R: 5’- TCCATTCCATCCAACGCC-3’
BioGrammatics wild type Pichia pastoris strain Bg10 was transformed with both expression constructs (Bg10 E-comp cells MP0002-BR002, ~200 ng of each vector DNA independently) (Transformation by electroporation in 1 mm cuvettes, 1150 Volts, 600 Ohms, and 10 uF capacitance). pJANaMF-CBM1-Bgluc transformants were selected on YPD-agar plates with Noursethricin (Nat, 100 ug/ml). pJAGaMF-6His-Bgluc transformants were selected on plates with G418 (800 ug/ml). Thousands of transformants were obtained for each construct. Cells from distinct colonies of greater than 200 transformants were picked from the lowest dilution selection plate and patched to a similar plate with the same selective drug (Nat or G418).
Initially, 96 well deep plates were used to screen the transformants, using the original Tre Bgl1 clone as a control; 4-methylumbelliferyl-β-D-cellobioside (MUC) was used to assay samples of each well. This growth format proved ineffective, as methanol induction failed to induce production of enzyme in any of the wells, including the control. Subsequently, individual clones were grown in 15 ml falcon tube cultures with daily additions of methanol. This screening method proved effective, demonstrating that essentially all clones produced active protein at similar levels. Clones were grown in 3 liter fermentors using BSM defined media 25. Cell mass was developed using glycerol as the primary feed for 24 hours, then switched to methanol to induce enzyme production. Fermentations were pH controlled and feed was added based on dissolved oxygen levels. Fermentations were run for four days on the methanol feed.The protein was purified using a method previously developed for purification of Bgl1 produced in fermentors using the MSU clone 24. The amino acid sequences of the two constructs (Bgl1H and Bgl1X) are shown in Figure 1. CBM1 (Bgl1X) and 6His-tag (Bgl1H) are shown in bold.
Figure 1.Protein sequences of Bgl1X and Bgl1H
Enzyme Purification and Properties
The two new strains produced more than 2-fold higher levels of protein than the MSU clone 24 in 3-liter fermentations. Purification of the new proteins was performed using the method developed for the MSU clone of Bgl1. After fermentation was complete, yeast cells were removed by centrifugation, and the clarified fermentation broths were concentrated using a Sartorius cross-flow ultrafiltration unit equipped with 20K cut-off membranes. After ultrafiltration, green colored material (alcohol oxidase microcrystals) was removed from the concentrate by cross-flow filtration through a 0.2 m membrane. The resulting material was concentrated and further purified by size exclusion chromatography on Sephacryl S100. This purification method was used to purify both BglH and Bgl1X, resulting in a purity of >95% for the two proteins (Figure 2). Attempts to purify either Bgl1H and BglX from centrifuged fermentation broth using Ni affinity chromatography or ion exchange chromatography was unsuccessful.
Figure 2.Legend Lane 1, MW standards, Lane 3-5 2, 5, and 10 mg purified Bgl1H, Lane 6-8, 2, 5, and 10 mg purified Bgl1H, Lane 9-11, 2, 5, and 10 mg purified Bgl1X.
Activity of Bgl1H on Disaccharide Substrates
The enzymatic activity of Bgl1H was investigated using 4-methylumbelliferyl and 2-nitrophenyl monosaccharides. As expected, the enzyme was active on both 2-nitrophenyl-β-D-glucopyranoside, 4-methylumbelliferyl-β-D-glucopyranoside (MUG). The specific activity of the enzyme was 11 u/mg at 50°C, pH 5.0, using 2-nitrophenyl-β-D-glucopyranoside as substrate. Bgl1H was also active on 4-methylumbelliferyl-β-D-xylopyranoside (MUX) and 2- nitrophenyl-β-D-xylopyranoside with a specific activity of 1.0 u/mg on 2- nitrophenyl-β-D-xylopyranoside. The enzyme showed no activity on the mannose or galactose-containing substrates. Hydrolysis of 2- nitrophenyl-β-D-glucopyranoside was inhibited by 10 mM glucose or cellobiose or 100 mM xylose. The inhibition by xylose confirms that a single active site is responsible for both hydrolysis of 2-nitrophenyl-β-D-glucopyranoside and other glucans and 2-nitrophenyl-β-D-xylopyranoside and other xylans. Assays of Bgl1X yielded specific activities statistically indistinguishable from the values obtained for Bgl1H, indicating the CBM1 module does not influence the rate on small substrates. Based on these results, Bgl1H was used for all kinetic studies.
Activity of Bgl1H on Polysaccharide Substrates
Previous work 15 reported on the ability of the enzyme to produce glucose from a number of low molecular weight substrates, with a clear specificity for β-(1,3)-laminarbiose 15. In this work, glucose production by Bgl1H was determined using an expanded range of substrates including cellodextrins, reduced cellodextrins, and polysaccharides. The rate of glucose production increased with the chain length of cellodextrin, (Table 1) with an approximate three-fold increase from cellobiose to cellohexaose. The reduced cellodextrins showed a similar behavior, with the rate of glucose formation from each reduced cellodextrin corresponding to the non-reduced cellodextrin with one fewer glucose unit. The rate of glucose production from laminarbiose (G3Gp) was similar to that of cellobiose (G4Gp). The rates of glucose production from the polysaccharide substrates were unusually high. The rate of glucose production from curdlan was approximately three-fold higher than from laminarbiose, and the rate of glucose production from β-glucan and lichenan was ten-fold higher than from cellobiose. These results indicate that the preferred substrates for Bgl1H are β-(1,3)- and β-(1,4)-polysaccharides and not laminarbiose or another disaccharide substrate as indicated previously.
Table 1. Rates of glucose formation by Bgl1H| Substrate | Structure | mmoles glucose/mg-min |
| β -(1,4)-glucans | ||
| cellobiose | G4Gp | 0.444 |
| cellotriose | G4G4Gp | 0.640 |
| cellotetraose | G4G4G4Gp | 0.850 |
| cellopentaose | G4G4G4G4Gp | 1.165 |
| cellohexaose | G4G4G4G4G4Gp | 1.252 |
| β -(1,4)-glucan alcohols | ||
| cellotriitol | G4G4GOH | 0.478 |
| cellotetraitol | G4G4G4GOH | 0.602 |
| cellopentaitol | G4G4G4G4GOH | 0.867 |
| cellohexaitol | G4G4G4G4G4GOH | 1.158 |
| Mixed β -(1,3)-(1,4)-glucans | ||
|---|---|---|
| b-glucan | G4GnG3G4GnG3Gp | 4.810 |
| lichenan | G4GnG3G4GnG3Gp | 4.870 |
| β -(1,3)-glucans | ||
| laminarbiose | G3Gp | 0.415 |
| curdlan | (G3G3)np | 1.282 |
The xylosidase activity of Bgl1H was evaluated using xylobiose, xylotriose, and xylotetraose. Hydrolysis of xylobiose by BGL1H was extremely slow (4% of the rate of cellobiose hydrolysis), but the hydrolysis rates increased rapidly with xylotriose and xylotetraose as substrates to approximately 25% of the rates of the corresponding cellobiosides (Table 2). Higher oligomers are not available, a result of the substitution on the xylose backbone of the xylan used for preparation of the oligoxylans.
Table 2. Rates of xylose formation by Bgl1H| Substrate | Structure | mmoles xylose/mg-min |
| xylobiose | X4Xp | 0.018 |
| xylotriose | X4X4Xp | 0.163 |
| xylotetraose | X4 X4X4Xp | 0.268 |
Hydrolysis of Glucans by Bgl1
The conversion of β-glucan to glucose by Bgl1H and four bacterial β-glucosidases with similar temperature and pH optima was determined (Table 3). Bgl1H showed 68% conversion of the β-glucan to glucose at 66 hours, and extended incubation resulted in 92% conversion of the β-glucan to glucose. The bacterial enzymes tested did not generate more than 2% glucose under the same conditions. Under the same conditions, Bgl1H produced 10.3% glucose from PASC, while the bacterial enzymes produced no significant amount of glucose. The bacterial β-glucosidases appear to be unable to digest either glucan polysaccharides, while Bgl1H is able to extensively degrade these partially soluble polysaccharides containing β-(1,4)-linkages, and mixtures of β-(1,4)-linkages and β-(1,3)-linkages, an activity not previously seen with this enzyme or other β-glucosidases.
Table 3. Degradation of glucans by b-glucosidases| Enzyme | β -Glucan Conversion | PASC Conversion |
| Blank | 0.0% | 0.1% |
| Bcel_0705 | 0.0% | 0.2% |
| Bcel_0282 | 0.2% | 0.0% |
| Aaz_81839 | 1.0% | 0.0% |
| Cthe_0212 | 1.8% | 0.0% |
| Bgl1H | 67.6% | 10.3% |
When evaluated using either of two insoluble celluloses, Whatman 1 filter paper or Avicel PH-10, Bgl1H and BglX produced no measurable glucose from these crystalline (insoluble) β-(1,4)-linked celluloses. Addition of Trichodermaendo- and exo-cellulases to Bgl1H results in almost complete conversion of these substrates to glucose 15.
When evaluated on insoluble β-(1,3)-linked curdlan, a maximum of approximately 20% hydrolysis to glucose was obtained using Bgl1H or Bgl1X, significantly better conversion than observed with insoluble crystalline or acid-swollen celluloses. Addition of a curdlanase, (Bcell_068326) to conversion reactions resulted in hydrolysis of approximately 70% of the curdlan to glucose. This increase in glucose production suggests crystalline regions within the curdlan may limit hydrolysis.
Xyloglucan differs from other glucans in possessing three out of four glucose residues substituted with α-(1,6)-linked xylose or substituted xylose residues. Even with extended incubations, Bgl1H produced less than 1% of the available glucose from tamarind xyloglucan. Addition of A. niger α-xylosidase (αXyl) 21, an enzyme capable of removing α-(1,6)-linked xylose from xyloglucan raised production of glucose to 4% of the available glucose (Table 4). Addition of an endo-acting xyloglucanase, Sfla_0771, or an endo-acting cellulase, Cthe_0797 increased glucose release to approximately 50% of the glucose present in the xyloglucan. Addition of the combination of a cellulase and xyloglucanase did not further increase glucose production.
Table 4. Degradation of xyloglucan by Bgl1H| Enzyme | Glucose produced |
|---|---|
| Bgl1H | 0.9% |
| +αXyl | 4.0% |
| +αXyl + Sfla_0771 | 47.7% |
| +αXyl + Cthe_0797 | 51.0% |
| +αXyl + Sfla_0771+ Cthe_0797 | 49.2% |
The enzyme did not release glucose from dextran, pullulan or starch, indicating an absolute specificity for β-linked glucan. Bgl1H produced no measurable glucose from glucomannan when incubated in the absence or presence of endo-mannanase, indicating the enzyme was unable to cleave glucose β-(1,4)-linked to mannose.
Hydrolysis of Arabinoxylooligosaccharides and Xylan
Incubation of arabinoxylooligosaccharides with Bgl1H resulted in no release of xylose, the result of α-2 and α-3-arabinose substitutions on the xylan backbone. When supplemented with α-arabinofuranoside, (G11MC16_1557), Bgl1H converted 29% of a 1.0% solution of xylooligosaccharides to xylose in 42 hours, a result superior to three of four thermostable bacterial xylosidases (Table 5). Prolonged incubation showed Bcel_0705 peaked at 42% conversion to xylose, while Tre BglA continued to convert the xylooligosaccharides, reaching 57% conversion.
Table 5. Conversion of Xylooligosaccharides to Xylose| Enzyme | XO Conversion |
|---|---|
| Blank | 1.2% |
| Bcel_0538 | 4.0% |
| Bcel_0385 | 1.3% |
| Bcel_0821 | 1.2% |
| Tre BglA | 29.6% |
| Bcel_0705 | 40.7% |
Bgl1 produced xylose from xylan when supplemented with xylanase (Cthe_2972) and α-arabinofuranoside (G11MC16_1557). Using 0.2% birchwood xylan, the combination of Bgl1H + xylanase + α-arabinofuranoside released 31% of the available xylose after 18 hr incubation, comparable to the 39% released using a combination of β-xylosidase (Y412MC61_2711) + xylanase + α-arabinofuranoside.
Hydrolysis of Glucan in Grain Samples
To determine if Tre Bgl1H could hydrolyze β-glucan and xylan present in whole grain samples, release of glucose and xylose were measured using commercial samples. Incubation for 115 hours resulted in significant formation of glucose in all samples, with barley and wheat flours being highest (Table 6). No glucose was formed in samples incubated identically without enzyme addition. Addition of thermostable β-glucanase (G11MC16_1587) and Bgl1 to an identical barley flour reaction resulted in production of 650 mg of glucose from 10g (6.5% by weight) of barley flour. The extractable β-glucan content of barley ranges from 1.8 to 5.4% 27, indicating the hydrolysis is able to convert all extractable and unextractable β-glucan to glucose.
Table 6. Formation of glucose from milling products| Milling Product | Glucose produced (mg) |
|---|---|
| Barley flour | 446 |
| Wheat flour | 362 |
| Wheat bran | 15 |
| Scottish oatmeal | 155 |
| Oat bran | 112 |
Discussion
This work reports the cloning, purification and first full characterization of Trichoderma reesei (Hypocreajecorina) Bgl1as both a his-tagged version and a version containing an N-terminal CBM1 module. The codon-optimized constructs yielded higher protein concentrations in the fermentation, and were significantly easier to purify than the original construct. Both forms were purified to greater than 95% purity, and both possessed essentially identical activities. The CBM1 module did not change either the rate of hydrolysis of tested substrates, and the CBM1 module did not improve conversion of crystalline or amorphous cellulose or other substrates. Bgl1 possesses a number of unique properties that are not found in other microbial β-glucosidases. The rate of Bgl1 increases with increasing saccharide chain length; most microbial β-glucosidases have maximum rates on disaccharides, with little or no activity on tetrasaccharides or higher polymers. The rates of glucose production from the polysaccharide substrates were unusually high. The rate of glucose production from curdlan was approximately three-fold higher than from laminarbiose, and the rate of glucose production from β-glucan and lichenan was ten-fold higher than from cellobiose. These results indicate that the preferred substrates for Bgl1H are β-(1,3)- and β-(1,4)-polysaccharides and not laminarbiose or another disaccharide substrate as indicated previously 15. Bgl1 also possesses β-xylosidase activity, again with a rate increasing with chain length.
The basis of the increasing rate with saccharide chain length is unclear. The x-ray structure (Figure 3) shows the active site in a cleft between two domains of the enzyme and clearly shows three substrate binding sites (-1. +1 and +2). No clearly define polysaccharide binding sites outside the active site are mapped, however, the crystal structure was not determined using a polysaccharide substrate. It is conceivable that this cleft binds to polysaccharides via a number of weak hydrophobic interactions or hydrogen bonds that keep the polysaccharide in the cleft and thereby increase the rate of hydrolysis via a processive mechanism 28. Additional structural and kinetic experiments are need to further clarify the action of this unique enzyme.
Figure 3.Crystal structure of Bgl1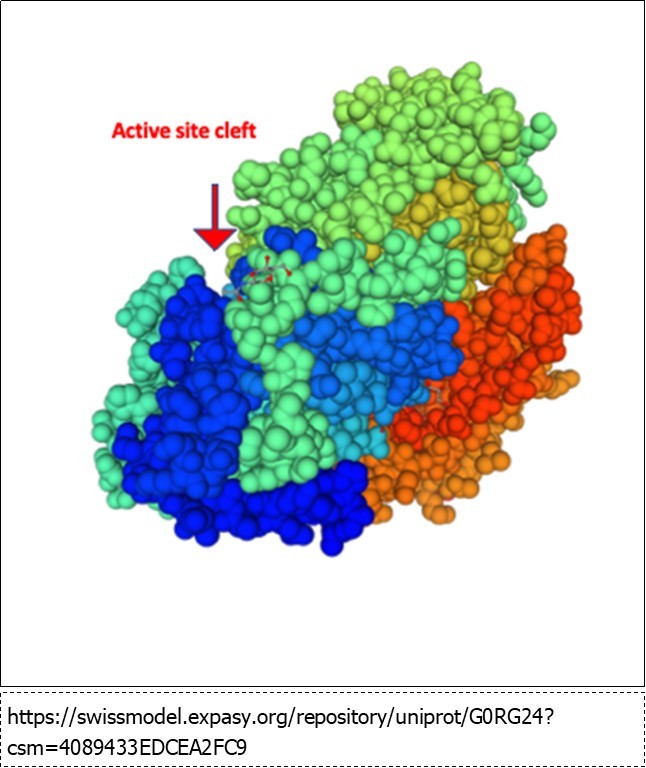
Conclusions
The rising cost of soy and corn has led to the use of other energy and protein sources in both hog and chicken feeds. A wide variety of materials can be utilized as energy feeds including barley, flax, oats, oat bran, rye, sorghum, triticale, and wheat 29. A problem common to many of these energy sources is the high level of soluble fiber present in the materials 30; this soluble fiber is primarily composed of xylan and β-glucan. The β-glucan content in whole grain oats ranges from 2 to 8% of dry weight; the β-glucan content of oat bran concentrate is higher, between 15 and 35% of dry weight. Other grains, especially barley, also possess significant amounts of β-glucan (Figure 4).
Figure 4.beta-glucan content of selected grains, data from 31.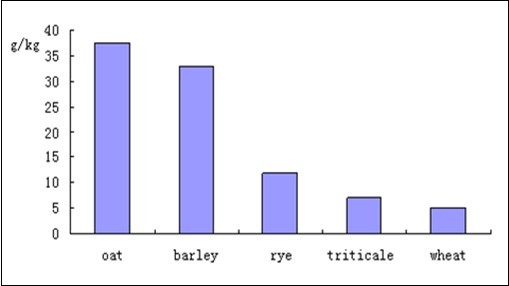
Present in a number of grains, beta-glucan is a homopolymer of glucose with predominantly beta- (1,4) linkages interspersed with beta- (1,3) linkages. The molecular weight of barley beta-glucan has been reported as 175,000 daltons 32 and 250,000 daltons The anti-nutritive effects of beta-glucan appear related to the same properties that have gained beta-glucan interest over its ability to improve glucose and insulin regulation and to lower blood cholesterol levels 33. Pigs fed diets high in fiber had higher maintenance energy requirements due the nutrient needs for increased viscera 34. Fiber was not degraded by the pig in the small intestine, but partially digested in the large intestine by microbes, resulting in increased formation of volatile fatty acids 35, 36. A number of endo-acting enzymes cleave beta-glucan including lichenanases 37 and endo-cellulases 31; these two classes of enzymes are the source of commercially available beta-glucanases. These enzymes generate cellobiose and cellotriose oligosaccharides with (1,3) linked glucose residues attached. These generated oligosaccharides cannot be utilized by the animals, but are converted to volatile organic acids by bacteria in the intestines. A preferred enzyme would be an exo-acting beta-glucanase, one that cleaves beta-glucan to produce glucose for assimilation by the animal. The results presented here suggest Bgl1 to be this enzyme, a single exo-acting beta-glucanase that can convert over 90% of beta-glucan to glucose. Bgl1 has significant potential to improve animal feed utilization as well as applications in other areas such as fermentation.
Authors’ Contributions
PB designed the study, analyzed the data, purified the enzymes and wrote the manuscript. DX performed all cellulose degradation studies. LA performed all fermentations. DM managed the enzyme synthesis and cloning. All authors read and approved the final manuscript.
Acknowledgements
This work was funded by the DOE Great Lakes Bioenergy Research Center (DOE BER Office of Science DE-FC02-07ER64494). The funding agency was not involved in the study design, collection, analysis and interpretation of data; or in the writing of the manuscript.
References
- 1.P J Brumm. (2013) Bacterial genomes: what they teach us about cellulose degradation. Biofuels. 4(6), 669-681.
- 2.Peterson R, Nevalainen H. (2012) Trichoderma reesei RUT-C30--thirty years of strain improvement. 158(1), 58-68.
- 3.Li Y. (2016) Overproduction of cellulase by Trichoderma reesei RUT C30 through batch-feeding of synthesized low-cost sugar mixture. , Bioresour Technol 216, 503-10.
- 4.Wang S. (2013) Enhancing cellulase production in Trichoderma reesei RUT C30 through combined manipulation of activating and repressing genes. , J Ind Microbiol Biotechnol 40(6), 633-41.
- 5.Li Z. (2015) Synergistic and Dose-Controlled Regulation of Cellulase Gene Expression in Penicillium oxalicum. PLoS Genet. 11(9), 1005509.
- 6.R D Brown. (1976) Structural characterization of a glycoprotein cellulase, 1,4-beta-D-glucan cellobiohydrolase C from Trichoderma viride. , Biochim Biophys Acta 446(2), 371-86.
- 7.Rouvinen J. (1990) Three-dimensional structure of cellobiohydrolase II from Trichoderma reesei. Science. 249(4967), 380-6.
- 8.Nakazawa H. (2008) Characterization of the catalytic domains of Trichoderma reesei endoglucanase I, II, and III, expressed in Escherichia coli. Appl Microbiol Biotechnol. 81(4), 681-9.
- 9.R H Bischof, Ramoni J, Seiboth B. (2016) Cellulases and beyond: the first 70 years of the enzyme producer Trichoderma reesei. Microb Cell Fact. 15(1), 106.
- 10.R L Mach. (1995) The bgl1 gene of Trichoderma reesei QM 9414 encodes an extracellular, cellulose-inducible beta-glucosidase involved in cellulase induction by sophorose. Mol Microbiol. 16(4), 687-97.
- 11.P V Harris. (2010) Stimulation of lignocellulosic biomass hydrolysis by proteins of glycoside hydrolase family 61: structure and function of a large, enigmatic family. Biochemistry. 49(15), 3305-16.
- 13.J A Langston. (2011) Oxidoreductive cellulose depolymerization by the enzymes cellobiose dehydrogenase and glycoside hydrolase 61. Appl Environ Microbiol. 77(19), 7007-15.
- 14.Leggio Lo, D Welner L, L De Maria. (2012) A structural overview of GH61 proteins - fungal cellulose degrading polysaccharide monooxygenases. , Comput Struct Biotechnol J 2, 201209019.
- 15.Karkehabadi S. (2014) Biochemical characterization and crystal structures of a fungal family 3 beta-glucosidase. Cel3A from Hypocrea jecorina. J Biol Chem 289(45), 31624-37.
- 16.Li C. (2016) A beta-glucosidase hyper-production Trichoderma reesei mutant reveals a potential role of cel3D in cellulase production. Microb Cell Fact. 15(1), 151.
- 17.Xue X. (2016) Revisiting overexpression of a heterologous beta-glucosidase in Trichoderma reesei: fusion expression of the Neosartorya fischeri Bgl3A to cbh1 enhances the overall as well as individual cellulase activities. Microb Cell Fact. 15(1), 122.
- 18.Dashtban M, Qin W. (2012) Overexpression of an exotic thermotolerant beta-glucosidase in trichoderma reesei and its significant increase in cellulolytic activity and saccharification of barley straw. Microb Cell Fact. 11, 63.
- 19.Walton J, Banerjee G, Car S.GENPLAT: an automated platform for biomass enzyme discovery and cocktail optimization. , J Vis Exp 2011(56).
- 20.Zhou S, L O Ingram. (2001) Simultaneous saccharification and fermentation of amorphous cellulose to ethanol by recombinant Klebsiella oxytoca SZ21 without supplemental cellulase. Biotechnology Letters. 23(18), 1455-1462.
- 21.J S Scott-Craig. (2011) Biochemical and molecular characterization of secreted alpha-xylosidase from Aspergillus niger. , J Biol Chem 286(50), 42848-54.
- 22.Y H Zhang. (2006) A transition from cellulose swelling to cellulose dissolution by o-phosphoric acid: evidence from enzymatic hydrolysis and supramolecular structure. Biomacromolecules. 7(2), 644-8.
- 23.Y S Kim, S J Yeom, D K Oh. (2011) Characterization of a GH3 family beta-glucosidase from Dictyoglomus turgidum and its application to the hydrolysis of isoflavone glycosides in spent coffee grounds. J Agric Food Chem. 59(21), 11812-8.
- 24.Banerjee G. (2010) Synthetic enzyme mixtures for biomass deconstruction: production and optimization of a core set. Biotechnol Bioeng. 106(5), 707-20.
- 25.C B Matthews. (2018) Development of a general defined medium for Pichia pastoris. Biotechnol Bioeng. 115(1), 103-113.
- 26.Mead D, Drinkwater C, P J Brumm. (2013) Genomic and enzymatic results show Bacillus cellulosilyticus uses a novel set of LPXTA carbohydrases to hydrolyze polysaccharides. PloS one. 8(4), 61131.
- 27.HAVRLENTOVÁ M, KRAIC J. (2006) Content of β-D-glucan in cereal grains. , Journal of Food and Nutrition Research 45(3), 97-103.
- 28.G T Beckham. (2014) Towards a molecular-level theory of carbohydrate processivity in glycoside hydrolases. Curr Opin Biotechnol. 27, 96-106.
- 30.Alternative Feed Ingredients in Swine Diets II: Use, Advantages and Disadvantages of Common Alternative Feedstuffs.
- 31.S M Tosh. (2004) Evaluation of structure in the formation of gels by structurally diverse (1‚3) (1‚4)-beta-d-glucans from four cereal and one lichen species. Carbohydrate Polymers. 57(3), 249-259.
- 32.Grimm A, Kruger E, Burchard W. (1995) Solution properties of B-D-(1,3)( 1, 4)-glucan isolated from beer. Carbohydrate Polymers. 27, 205-214.
- 33.P J Wood. (2004) Relationships between solution properties of cereal B-glucans and physiological effects—a review. Trends in Food Science and Technology 15, 313-320.
- 34.C M Grieshop, D E Reece, G C Fahey. (2001) Nonstarch polysaccharides and oligosaccharides in swine nutrition. in Swine Nutrition, Second Edition, A.J. Lewis 107-130.
- 35.Knudsen Bach, E K, Hansen I. (1991) Gastrointestinal implications in pigs of wheat and oat fractions. 1. Digestibility and bulking properties of polysaccharides and other major constituents. The British journal of nutrition. 65(2), 217-32.
- 36.Knudsen Bach, E K. (1991) Gastrointestinal implications in pigs of wheat and oat fractions. 2. Microbial activity in the gastrointestinal tract. The British journal of nutrition. 65(2), 233-48.
- 37.P J Wood, Weisz J, B A. (1991) Molecular characterisation of cereal B-D-glucans. Structural analysis of oat B-D-glucan and rapid structural evaluation of B-D-glucans from different sources by high- performance liquid chromatography of oligosaccharides released by lichenase. Cereal Chemistry. 68, 31-39.