Abstract
Parachordoma is an extremely exceptional, peripherally situated soft tissue neoplasm arising at non axial locations. Also designated as soft tissue “myoepithelioma” or “mixed tumour”, the tumefaction histologically simulates a chordoma of axial skeleton and was referred to as “central chordoma” emerging within non- axial sites. Nevertheless, a distinctive immune profile categorises the lesion as a unique entity.
Author Contributions
Academic Editor: Pietro Scicchitano, Cardiology Department, Hospital of Ostuni (BR) - Italy.
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2020 Anubha Bajaj
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Preface
Parachordoma is an extremely exceptional, peripherally situated soft tissue neoplasm arising at non axial locations. Also designated as soft tissue “myoepithelioma” or “mixed tumour”, the tumefaction histologically simulates a chordoma of axial skeleton and was referred to as “central chordoma” emerging within non- axial sites. Nevertheless, a distinctive immune profile categorises the lesion as a unique entity. Principally described by Laskowski in 1951 as “chordoma periphericum”, the neoplasm was subsequently nomenclated as “parachordoma” by Dabska in 1977.
Parachordoma as a terminology is applicable on account of occurrence of vacuolated tumour cells 1, 2. Parachordoma is an indolent, gradually evolving neoplasm with occasional delayed tumour reoccurrence, infrequent metastasis and occasional mortality. Parachordoma can be contemplated as a potentially aggressive, low grade sarcoma with properties of localized tumour infiltration. Often misinterpreted, parachordoma can be adopted as a synonym for soft tissue neoplasms such as chondrosarcoma, extra- axial chordoma (EAC) or low grade sarcoma 3.
Disease Characteristics
Majority of parachordomas are benign, around 14.5% instances relapse whereas 12.9% neoplasms are metastatic. Parachordoma demonstrates a male predilection with a male to female ratio of 1.3:1. Average age of disease emergence is 34.4 years although the neoplasm can arises between 4 years to 86 years 3.
Proportionate tumour incrimination is lower extremity (51.2%), upper extremity (26.8%), thorax, trunk and pelvis (19.5%), head and neck (15%) or buttocks (6.4%). Parachordoma of head and neck can appear in intracranial region, parietal skull or nares. Groin and gastric mucosa can exhibit a parachordoma 3, 4.
Delayed tumour reoccurrence is denominated in around 0.2% subjects and neoplastic reappearance occurs between 3 months to 12 years with tumour relapse at an average of 3.8 years following surgical resection. As the neoplasm is of uncertain lineage, it may be scripted as a myoepithelioma, myoepithelial carcinoma or mixed tumour and is accompanied by pertinent, confirmed biological behaviour. Genesis of parachordoma is attributed to ectopic rests of notochord, Schwann cells, myoepithelial cells or specialized synovial cells although convincing evidence regarding a specific cellular lineage is absent 4, 5. Myoepithelial carcinoma is an exceptional neoplasm, preponderant within paediatric population, which can reoccur and metastasize in an estimated 50% of histologically aggressive tumours 4.
Clinical Elucidation
Parachordoma is an indolent, painless, gradually progressive neoplasm and appears minimally aggressive as compared to chordoma. Parachordoma predominantly occurs within deep-seated soft tissue of distal extremities such as deep fascia, skeletal muscle, tendon, synovial tissue or soft tissue abutting the bone 3, 4.
Parachordoma is associated with secondary manifestations, tumour compression, pain or restriction of anatomical functions.
Parachordoma can demonstrate tumour metastasis of chordoma- like sarcoma with pulmonary deposits. Brain metastasis can occur. Tumours associated with widespread metastasis and localized tumour reoccurrence can engender tumour mortality. Chest wall parachordoma can metastasize to lymph nodes. Metastatic tumour is extensively cellular and pleomorphic. Fatality can ensue following surgery, chemotherapy or pertinent radiotherapy 5.
Histological Elucidation
Parachordoma morphologically simulates a chordoma although tumour countenance is variable. Grossly, parachordoma depicts a firm, lobulated or nodular appearance, variable magnitude of 2 centimetres to 8 centimetres with a mean diameter of 3.5 centimetres. Tumour perimeter is well defined. Cut surface is greyish/ white, gelatinous, semi-translucent with cartilaginous zones and disseminated foci of myxoid material 5. Necrosis and haemorrhage may be absent although enlarged neoplasms are associated with focal haemorrhage, necrosis and cystic degeneration. Cogent tissue specimen obtained exhibits a tumour composed of nests and cords of cells with eosinophilic, clear, vacuolated cytoplasm and uniform, spherical to elliptical, hyperchromatic or bland nuclei. The neoplasm displays a reticular or trabecular pattern of evolution, intermingled with foci of myxoid, cartilaginous or hyalinised matrix.
Microscopically, a well circumscribed, un-encapsulated, multinodular neoplasm comprised of spheroidal, epithelioid or plump spindle- shaped cells is denominated with tumour cells configuring clusters, chains, nodules and whorls although a glandular architecture is typically absent. An encompassing chondromyxoid, hyaline stroma is discerned and the tumour is traversed with fibrous tissue septa 5, 6.
Three distinct cellular subtypes constitute a parachordoma. Epithelioid cells, miniature glomoid cells and spindle-shaped cells. Besides, the neoplasm has a distinctive population of cells with vacuolated cytoplasm, simulating physaliferous cells of chordoma 5.
Parachordoma is composed of aggregates, whorls and a pseudo-glandular pattern of spheroidal cells embedded within a focally myxoid, hyaline stroma, subdivided by broad, fibrous tissue septa. Foci of fresh and old haemorrhage are exemplified. Tumour necrosis or vascular invasion is usually absent. Mitotic activity is minimal and usually below < one mitosis per 20 high power fields5, 6.
Multinodular tumour architecture is constituted by miniature, uniform, elliptical or spindle-shaped cells with scanty, eosinophilic, finely vacuolated cytoplasm and miniature, regular or hyperchromatic nuclei. Myxoid tumour matrix is envelops neoplastic cells configuring cords and strands. Nuclear pleomorphism is absent. Tiny foci of necrosis with focal chondroid differentiation may ensue6. Meningo-endothelial cells, physaliferous cells, rhabdoid cells, ductal differentiation and anaplastic cellular dedifferentiation is usually absent. Myoepithelial carcinoma displays nuclear atypia, elevated mitotic rate and extensive foci of tumour necrosis5. Figure 1, Figure 2, Figure 3, Figure 4, Figure 5, Figure 6, Figure 7, Figure 8.
Figure 1. Parachordoma on fine needle aspiration cytology depicting clusters of spheroidal cells with eosinophilic cytoplasm, regular nuclei and clumped, encompassing myxoid stroma 13.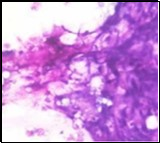
Figure 2. Parachordoma delineating clusters of vacuolated, spherical cells with acidophilic cytoplasm, uniform nuclei and an immune non reactive CD34- 13.
Figure 3. Parachordoma exemplifying clusters and nests of spherical, vacuolated cells with abundant, acidophilic cytoplasm, regular nuclei and an enveloping cartilaginous stroma 14.
Figure 4. Parachordoma depicting cords and aggregates of vacuolated cells with acidophilic cytoplasm and regular, miniature nuclei with an encompassing myxoid stroma 15. 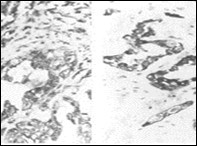
Figure 5. Parachordoma exhibiting nests and clusters of spheroidal cells with eosinophilic, vacuolated cytoplasm, uniform, bland nuclei and a circumscribing myxoid stroma 15. 
Figure 6. Parachordoma delineating clusters and nests of spindle-shaped and spherical, vacuolated cells with eosinophilic cytoplasm and uniform nuclei 16.
Figure 7. Parachordoma depicting immune reactivity to S100 protein 16.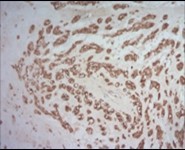
Figure 8. Parachordoma demonstrating immune reactivity to vimentin 16.
Immune Histochemical Elucidation
Parachordoma is immune reactive to cytokeratin CAM 5.2 and glial fibrillary acidic protein (50%). A subset of tumour cells are immune reactive to CD99 and smooth muscle actin (SMA). Stroma is intensely highlighted with an alcian blue stain wherein staining intensity is reduced following hyaluronidase digestion. Tumour cells are immune reactive to epithelial membrane antigen (EMA), CK8/18, S100 protein, calponin (50%), vimentin, type IV collagen and CD117 6.
Tumour cells are immune non reactive to CK1/10, cytokeratin AE1/AE3, CK20, CK7, CK19, CK12-17,smooth muscle actin (SMA), muscle specific actin (MSA), carcino-embryonic antigen (CEA), desmin, CD34, CD31, CD10, glial fibrillary acidic protein (GFAP), p63, TFE3, melan -A, brachyury, Human Melanoma Black-45 (HMB-45), D2-40 and α- inhibin.
Type IV collagen is immune reactive, envelops clusters of tumour cells and articulates a nest- like appearance 6, 7.
Differential Diagnosis
Upon employment of imaging techniques, parachordoma requires a segregation from diverse benign and malignant neoplasms such as malignant fibrous histiocytoma, fibrosarcoma or metastatic malignancies 6. Demarcation is required from metastatic mucinous adenocarcinoma, intramuscular myxoma and chondroid parachordoma6, 7.
Histological differentiation is mandated from renal cell carcinoma, chondroid lipoma, myxoid liposarcoma, adjunctive renal neoplasms, extra skeletal myxoid chondrosarcoma, chordoma and pleomorphic adenoma of salivary gland 6, 7.
Renal cell carcinoma is a clear cell carcinoma of adult age group typically delineating an extraneous golden tinge on account of lipid rich cellular component. The neoplasm depicts focal necrosis and haemorrhage. Nests of clear cells are segregated by a vascular framework 7.
Clear cell renal cell carcinoma is immune reactive to cytokeratin and CD10.
Papillary, mucinous, tubular and spindle cell renal cell carcinoma depict a prominent tubular architecture7.
Chromophobe cell carcinoma delineates an admixture of clear and eosinophilic cells and may be associated with chromosomal translocation Xp11.2 /TFE3 besides gene fusion renal cell carcinoma(Xp11.2 RCC) with expression of TFE37.
Collecting duct carcinoma characteristically exhibits an infiltrative border, tubulo- papillary architecture and marked cytological atypia.
Medullary renal cell carcinoma enunciate tubular articulations with neutrophilic infiltration and rhabdoid morphology6, 7.
Sarcomatoid renal cell carcinoma is comprised of spindle shaped cells although mitotic figures are exceptional, as denominated in benign or low grade neoplasms.
Benign and malignant renal tumours such as adult renal myoepithelial hamartoma requires differentiation.
Metanephric adenoma is composed of low, cuboidal tumour cells with basophilic cytoplasm and infrequent tubular articulations. Encompassing myxoid stroma is absent. Renal cell carcinoma and associated renal neoplasms can be eliminated with pertinent clinical, microscopic and immune histochemical evaluation6, 7.
Chondroid lipoma delineates a distinctive component of mature lipoblasts. The neoplasm is immune reactive to S100 protein and is immune non reactive to cytokeratin (CK).
Myxoid liposarcoma displays a distinct morphology and the adipose tissue component can be emphasized with Sudan III stain. Also, FUS- CHOP genetic fusion with break –apart chromosomal rearrangement is discernible 6, 7.
Parachordoma necessitates distinction from conventional chondrosarcoma and extra skeletal myxoid chondrosarcoma as the neoplasms are associated with significant localized tumour reoccurrence and distant metastasis, in contrast to benign parachordoma which exhibits exceptional tumour relapse or metastasis6, 7.
Extra skeletal myxoid chondrosarcoma emerges in middle aged or elderly individuals. A lobular or nodular tumour pattern is discerned with tumour parenchyma subdivided by fibrous tissue septa. Tumour nodules are solid, tumour cells are spherical or plump, spindle-shaped and are imbued with pale cytoplasm and hyperchromatic nuclei with inconspicuous nuclei. Vacuolated or miniature, spheroidal tumour cells are absent. Encompassing stroma is uniform and composed of mucopolysaccharides which is resistant to digestion by hyaluronidase and can be suitably stained with alcian blue 6, 7.
Extra skeletal myxoid chondrosarcoma lacks differentiation towards epithelial or myoepithelial cells. Extra skeletal myxoid chondrosarcoma is immune reactive to vimentin and desmin. Few instances are immune reactive to S100 protein, cytokeratin or epithelial membrane antigen (EMA) although diffuse immune reactivity to CAM 5.2 or S100 protein is absent. Around 75% of extra skeletal myxoid chondrosarcomas characteristically display a balanced chromosomal translocation t(9;22)(q22;q12) with break point of EWS gene situated upon chromosome 22q21 and CHN gene located upon chromosome 9q22. Also, EWSR1 gene break-apart chromosomal rearrangement can be discerned with fluorescent in situ hybridization (FISH) 7, 8.
Chordoma is consistently located within the axial skeleton, especially sacrococcygeal region, sphenocciptal region or vertebral column. Chordoma is comprised of vacuolated cells which stain intensely with alcian blue and are resistant to digestion with hyaluronidase. Chordoma is immune reactive to cytokeratin such as CK1/10, CK7, CK20, CK19, CK12-17 and immune non reactive to type IV collagen 7, 8.
In contrast, parachordoma is composed of diverse architectural patterns and immune profile. Parachordoma is predominantly discerned within deep-seated soft tissue of distal extremities as fascia, skeletal muscle, tendon, synovium or soft tissue abutting the bone. Morphologically, parachordoma is composed of vacuolated, spindle-shaped or spheroidal epithelial cells. Myxoid matrix demonstrates enhanced concentration of hyaluronic acid which stains intensely with alcian blue and is sensitive to digestion with hyaluronidase wherein tumour cells are devoid of alcian blue staining. Parachordoma is intensely immune reactive to type IV collagen and immune non reactive to CK7, CK19 and brachyury7, 8.
The neoplasms are immune reactive to cytokeratin CAM5.2 or cytokeratin 8/18, epithelial membrane antigen (EMA), vimentin and S100 protein 8.
Peripheral chordoma, immune reactive to brachyury, is disparate from parachordoma or myoepithelioma. Brachyury immune reactive, extra-axial chordoma incurs localized bone destruction and demonstrates a tendency to progress and reappear whereas parachordoma is minimally aggressive and delineates a favourable outcome8, 9.
Pleomorphic adenoma of the salivary gland demonstrates morphological diversity. Tumour components are epithelial, myoepithelial and stromal or mesenchymal cells. Epithelial components are denominated by various cell types such as squamous, spindle-shaped and clear cells. Mesenchymal cells are comprised of myxoid, cartilaginous or hyaline cells. Pleomorphic adenoma is immune reactive to cytokeratin, vimentin, CD10, S100 protein and calponin. Clinical assessment, morphological and radiographic features and cogent immune reactions can be adopted to differentiate primary pleomorphic adenoma/ carcinoma from parachordoma. Lymph node metastasis is documented in pleomorphic adenoma9.
Investigative Assay
Although non specific, imaging characteristics are crucial in evaluating the anatomy and diagnostic parameters of parachordoma.
Tumour staging mandates computerized tomography (CT) of the thoracic, abdominal and pelvic cavity. Computerized tomography (CT) depicts an enlarged extra axial neoplasm associated with erosion of abutting bone. Foci of calcification demonstrate a minimal signal intensity upon computerized tomography (CT). A heterogeneous pattern of contrast enhancement with zones of non enhancing cystic or necrotic modifications can be delineated. Restricted diffusion within the tumefaction upon diffusion weighted imaging or oedema of encompassing soft tissue is absent. Intra- tumoural and peri-tumoural vascular flow voids are normal10.
Magnetic resonance imaging (MRI) delineates non specific features of an enlarged, deep-seated, heterogeneous soft tissue mass, appearing as an enlarged tumefaction compressing circumscribing soft tissue along with bone erosion. The tumour is uniformly hyper-intense upon T2 weighted imaging and displays intermediate to minimal intensity upon T1 weighted imaging10.
Parachordoma demonstrates trisomy 15, loss of chromosome 1,16, 17, 9,10, 20 and 22 besides a structural rearrangement of chromosome 3 (del3q) and genomic translocation between chromosome 2 and 4 t (2p;4q), preponderantly discerned within a recurrent neoplasm.
Fluorescent in situ hybridization (FISH) studies demonstrate a lack of genomic rearrangements for EWSR1, NR4A3, EWS, FUS, SMARCB1 and PHF-1 genes, features which are concordant with primary myoepithelial lesion such as parachordoma11.
Therapeutic Options
Contemporary therapy of the locally infiltrative parachordoma is extensive surgical extermination of the lesion. Comprehensive surgical excision of the tumefaction with resection of a broad, perimeter of normal uninvolved tissue along with the biopsy tract is recommended. An intraoperative frozen section can be suitably adopted to assess tumour margin12. The benign myoepithelioma is associated with tumour relapse in around 20% instances and necessitates localized surgical excision with a healthy margin of tumour-free tissue. Adjuvant therapy is indicated in instances associated with metastasis11, 12. Extensive post operative monitoring is required as the essentially benign parachordoma may delineate tumour reoccurrence or metastasis within 3 months to 12 years11, 12.