Abstract
Theobjective of reviewing Hairy Cell Leukaemia may be achieved by emphasising the condition as a category of chronic lymphocytic leukaemia with hair like protrusions of the cytoplasm situated on the aberrant B cell surface. An infrequent disorder, hairy cell leukaemia contributes an estimated 2% of lymphoid malignancies with a male predominance ( M:F ::4-5:1). A majority (90%) of instances depict a mutant immunoglobulin heavy chain variable region (IGHV). The haematopoietic stem cells (HSCs) elucidate a B raf proto-oncogene( BRAF V600E gene- 7q34). An enlarged spleen may be discerned along with pancytopenia as a presenting symptom. The morphology of specific hairy cell leukaemia may be on account of an in vitro interaction of primary hairy cells with BRAF genes and MEK inhibitors, which incite a prominent MEK - ERK dephosphorylation, thereby curtailing transcriptional outpourings of the RAS- RAF- MEK-ERK pathway. Bone marrow aspiration or bone marrow trephine biopsy may be inadequate for diagnosis in 30%-50% individuals on account of extensive fibrosis and the bone marrow sections depict a characteristic interstitial infiltration of leukaemia cells.. Reticulin stains exhibit broad, dense reticulum fibres surrounding the individual or aggregates of leukaemia cells with fibrotic extensions into the abutting bone marrow. The immune reactivity of classic hairy cell leukaemia is concurrent CD19+ CD20+,CD 11c+, CD25+, CD103+ and CD123+. Immune staining for CD20+, annexin 1 and VE1 (a BRAF V600E stain) validates the diagnosis and analyses the extent of malignant bone marrow infiltration. Application of inhibitors of BRAF V600E gene is efficacious in patients resistant to standard therapy. An enlarged spleen beyond 3 centimetres of the left costal margin, a white blood cell count greater than 10000 cells/µL , circulating hairy cells in the peripheral blood greater than 5000 cells/µL and a β 2 micro-globulin level exceeding twice the normal range of 3 µg/ml delineate an inferior outcome with resistance to purine analogues (PNAs). CD38+ elucidation ensures a worse prognosis as does the lack of an IGHV mutation with a reduced overall survival,. A lack of BRAF genetic mutation in 10% -20% of hairy cell leukaemia comprises of inferior prognosis.
Author Contributions
Academic Editor: Pietro Scicchitano, Cardiology Department, Hospital of Ostuni (BR) - Italy.
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2018 Anubha Bajaj
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Preface
Originally delineated in 1958 by Bouroncle and colleagues, hairy cell leukaemia (HCL) is a category of chronic lymphocytic leukaemia with the terminology based on the hair like protrusions of cytoplasm situated on the surface of aberrant B cell 1. Hairy cell leukaemia contributes roughly 2% to the gamut of lymphoid malignancies with an estimated instances of 1000/year in the developed world. The leukaemia demonstrates a male predominance (M:F ::4-5:1)2. Hairy cell leukaemia has subsequently been established as an explicit entity by the world health organization(WHO) in the designated classifications of haematological and lymphoid neoplasm in 2008 /2016 3.
Cellular Origin of Hairy Cell Leukaemia
The delayed activation of post germinal centre memory B cells probably along with marginal zone B cells of the spleen may constitute a cellular origin of hairy cell leukaemia. A majority (90%) of instances may depict a mutant genetic profile of immunoglobulin heavy chain variable region (IGHV). The haematopoietic stem cells (HSCs)and the cellular component of Langerhans Cell Histiocytosis(LCH) and Erdheim Chester Disease (ECD) may elucidate a B raf proto-oncogene( BRAF V600E gene- 7q34). Majority of the mutant alleles accompanying these disorders may be situated in the CD14+ classical monocytes, the CD 16+ non classical monocytes and CD1c+ myeloid dendritic cells, located in the peripheral blood2. The mutant alleles may be dispersed within the HSCs and myeloid progenitors of the bone marrow. In hairy cell leukaemia the mutant alleles may be lacking within monocytes and myeloid cells, although may appear in the normal B lymphocytes and natural killer (NK) cells2.
Clinical Representation
The patients may display systemic symptoms such as fatigue , infection, splenomegaly besides a constitutional depiction of pancytopenia. An enlarged spleen may be discovered in a majority (90%) of the instances, although the feature may be infrequent in recent times, on account of the earlier discernment of the disorder. Pancytopenia may be the presenting symptom2, 4. The preliminary assessment of disease may mandate a differential leukocyte count with a reappraisal of the peripheral blood smear. Monocytopenia, when delineated, may prove to be a sensitive and specific indicator of hairy cell leukaemia. The appearance of leukaemia cells per se may be exceptional2.
Microscopic Elucidation
The morphology of hairy cell leukaemia may be specific , in contrast to the variants, on account of an in vitro interaction of the primary hairy cells with BRAF genes and MEK inhibitors, which may incite a prominent MEK - ERK dephosphorylation, thereby curtailing transcriptional outpourings of the RAS- RAF- MEK-ERK pathway. The particular occurrence may induce a deficit in hairy cell leukaemia specific genetic profile signature that may convert the morphology of hairy cells into smooth cells with ultimate apoptosis2. The objective of B actin and leukocyte specific transcript1 ( LST 1) in the establishment of a hairy cell morphology may be uncertain2.
The classic hairy cell is medium sized with a magnitude of 10-14µm. The moderately abundant or variable cytoplasm may be transparent or mildly basophilic. The cellular surface with the characteristic serrated perimeter depicts innumerable fragile or stout extensions of cytoplasm ,particularly discernible on the phase contrast and electron microscopy. The cytoplasm may exhibit vacuoles with occasional azurophilic granules4. The nucleus may be elliptical or reniform, folded or indented with a coarse, reticulated or a finely dispersed chromatin and inconspicuous nucleoli along with infrequent mitosis. Bone marrow aspiration or bone marrow trephine biopsy may be inadequate for diagnosis in 30%-50% individuals4. The trephine sections of the bone marrow may depict a characteristic interstitial pattern of leukaemic infiltration. Generally the bone marrow is hyper-cellular, though it may be hypo-cellular in 10-15% individuals4. The leukaemia cell ingress may be diffuse or partial, although diffuse infiltration is frequent. The partial variety of leukaemic dissemination may be ineptly categorized with an indeterminate differentiation from the uninvolved marrow. The malignant insertions may initially emerge as miniature, undefined, cellular loci. The formalin fixed, paraffin embedded sections may elucidate a crystalline zone or a “halo” appearance of the cells with a circumscribed nucleus on account of the plentiful cytoplasm4. The cellular margins may be intertwined. Fixation of bone marrow smears with Zenker’s fixative may demonstrate a retracted cytoplasm of the hairy cells with a consequent disconnected structure. The bone marrow in the absence of a malignant process may be hypo-cellular or hyper-cellular. Reticulin stains may delineate an enhanced accrual of broad, dense reticulum fibres surrounding the aggregates of leukaemia cells with the fibrous circumlocution of individual malignant cell and fibrotic extensions into the abutting, uninvolved bone marrow4.
The leukaemia cells may enunciate a characteristic immune phenotype, crucial for a confirmatory diagnosis. The peripheral blood mononuclear B cell population may display a kappa or lambda light chain restriction. The phenotype of classic hairy cell leukaemia may be delineated by concurrent, immune reactive CD19+ CD20+,CD 11c+, CD25+, CD103+ and CD123+. An intensely immune reactive CD200+ and a non reactive CD27- antigen may be present2, 4. Evaluation of a trephine bone marrow biopsy and bone marrow aspirate may define the degree of tumour infiltration. A dry tap on account of prominent bone marrow fibrosis may be elucidated at preliminary diagnosis. A decline in the normal haematopoiesis may account for a hypo-cellular marrow in 10% instances. Gradation of cellular infiltrating of the leukaemia within the bone marrow may be appropriately investigated with immune –histochemical stains2, 4. Immune staining for CD20+, annexin 1 and VE1 (a BRAF V600E stain] may validate the diagnosis and precisely analyse the extent of malignant bone marrow infiltration[8]. Determination of BRAF V600E mutation may be critical in therapeutically non responsive individuals with applicable standard therapy or in instances of multitudinous reoccurrences[9]. Deploying inhibitors of BRAF V600E gene may be efficacious in patients impervious to approved therapy. The mutation necessitates a comprehensive scrutiny of the implicated individuals with a sensitive molecular assay which may discern up to < 10% of the hairy leukaemia cells appearing in the peripheral blood smears or bone marrow aspirates diluted with peripheral blood or aspirates elucidating a dry tap[2,4]. Allele specific polymerase chain reaction (PCR) or a next generation sequencing may be optimally employed to circumvent false negative outcomes. If the leukaemia cells are sparse or if particularly sensitive & efficacious molecular techniques are not accessible, the application of appropriate immune histochemical stains to the bone marrow biopsy such as a BRAF V600E mutation stain (VE1) may detect the hairy cells and conclusively diagnose the condition[2,4,10]. Figure 1, Figure 2, Figure 3, Figure 4, Figure 5, Figure 6, Figure 7, Figure 8, Figure 9, Figure 10, Figure 11, Figure 12, Figure 13, Figure 14.
Figure 1. HCL: hairy cells infiltrating designated spaces(17).
Figure 2. HCL: hairy cells with projecting cytoplasm abutting bony trabaculae(18).
Figure 3. HCL: hairy cells dispersed within the native architecture(19).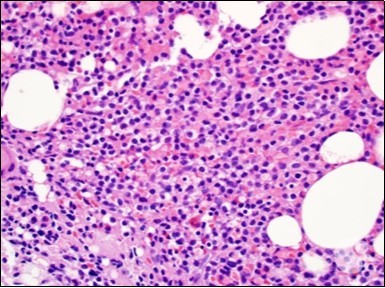
Figure 4. HCL: hairy cells with widely spaced nuclei(20).
Figure 5. HCL: hairy cells with infiltration in the spleen(21).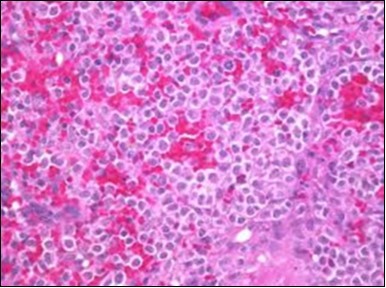
Figure 6. HCL: disseminated hairy cells with a clear cytoplasm(22).
Figure 7. HCL: blebs on the cellular surface with fine nuclear chromatin(23).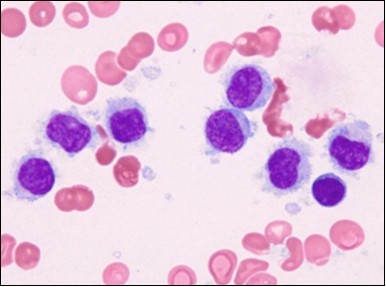
Figure 8. HCL: widely disseminated hairy cells within the bone marrow trabaculae(24).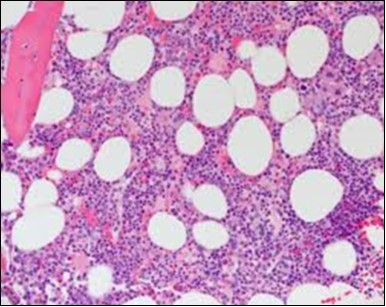
Figure 9. HCL: hairy cells within a bone marrow trephine biopsy(25).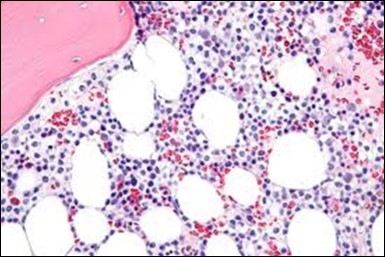
Figure 10. HCL: inconspicuous nucleoli, open-ended chromatic and surface protrusions(26). 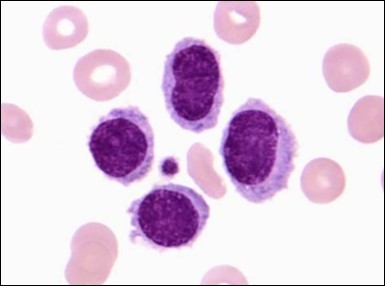
Figure 11. HCL Oral mucosa with soft tissue infiltration of hairy cells(27).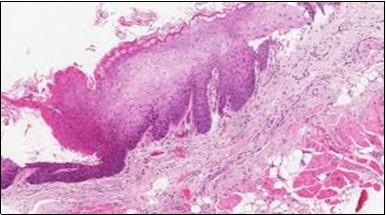
Figure 12. HCL: hairy cells with broad and fine projections of the cytoplasm(28).
Figure 13. HCL: hairy cells immune reactive for CD 11c(24).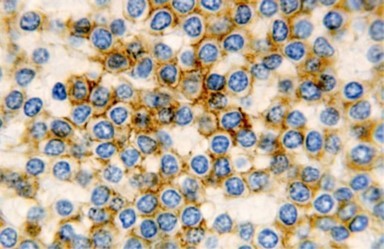
Figure 14. HCL: hairy cells with demonstrable tartrate resistant acidic phosphatise( TRAP) stain(29).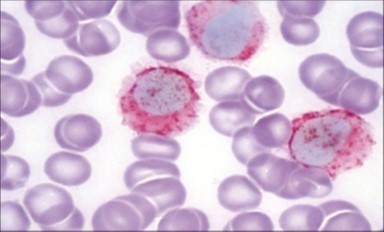
Disease Characteristics
A complete blood count with an assiduous peripheral smear may assist the detection of hairy cells. Characteristic immune phenotype of the B cell leukaemia clone may depict an augmented, intense staining of CD19 +, CD20+, CD22+ with CD200+. Malignant hairy cells may be non reactive or weakly reactive for CD5-,CD23-,CD10-, CD79b-, and CD27- though reactive for CD11C+, CD103+, CD123+ and CD25+11. An immunological scoring system of one point each to the manifested CD11C+,CD103+, CD123+ and CD25+ immune phenotypes may be proposed2. Majority (98%) of the hairy cell leukaemia depict a quantification of 3-4 whereas the variants of HCL demonstrate a magnitude of 0-1. According to the international consensus guidelines, a bone marrow trephine biopsy and/or a bone marrow aspiration may adequately configure the extent of the tumour infiltration besides discerning complicated disease (utilizing immune stains for CD20, CD76 and Annexin A 1)2, 4, 12.
Discordant Diagnosis
Hairy cell leukaemia necessitates a distinction from adjunctive disorders such as the hairy cell leukaemia –variant ( HCL –V) and splenic diffuse red pulp lymphoma (SDRPL)2. The hairy cell leukaemia variant (HCL-V) may incorporate a mere 10% of the instances of HCL and as yet remains a conditional form of leukaemia. The disseminated, aberrant lymphoid cells may elucidate an intermediary morphology betwixt a pro-lymphocyte and a hairy cell. Immunological quantification of the HCL-V for aforementioned parameters may be minimal (0-1). An absence of immune reactive CD25- and CD200- may manifest with an inconsistent or poorly enunciated CD1232.The individuals may lack mono-cytopenia. Likewise, the splenic diffuse red pulp lymphoma (SDRPL) may be cogitated as a provisory condition,though dissimilar from HCL-V. Majority (60%) of the small to medium sized, aberrant villous lymphoid cells appear in the peripheral blood and may depict a polar villous disposition with a miniature to absent nucleolus. The monoclonal B cells in the majority (97%)of the implicated SDRPL may exhibit an immune reactive CD11C+ with a discordantly demonstrated CD103+ (38%) and an occasional elucidation of CD123+ (16%) with CD25+ ( 3%)2.
Prognostic Indicators
An enlarged spleen beyond 3 centimetres of the left costal margin, leucocytosis with a white blood cell count greater than 10000 cells/µL , circulating hairy cells in the peripheral blood greater than 5000 cells/µL and a β 2 micro-globulin level exceeding twice the normal range of 3 µg/ml may be accompanied by an inferior therapeutic outcome and an emerging resistance to purine analogues (PNAs)2. Simulating the chronic lymphocytic leukaemia , an elucidation of CD38+ may ensure a worse prognosis6. The mutation of immunoglobulin heavy chain variable region (IGHV) gene may dictate prognostic assumptions of the disorder. Individuals lacking an IGHV mutation may depict a reduced overall survival, in contrast to the patients demonstrating the IGHV mutation. An estimated 40% of the hairy cell leukaemia variant( HCL –V) and 10% of classic hairy cell leukaemia may elucidate an IGHV 4-34 immunoglobulin heavy chain variable gene rearrangement13. VH 4- 34 reactive instances may determine a contemporary variant and subset of hairy cell leukaemia with an unfavourable prognosis, an elevated initial disease encumbrance, inadequate response to recognized therapy, a decline in the overall survival and an absence of BRAF V600E mutation2.
Genetic Eventualities
The employment of whole exome sequencing (WES) for the detection of a BRAF V600E somatic mutation may be advocated. The B raf proto-oncogene( BRAF gene- 7q34) comprises of 18 exons and a genetic mutation of exon 15 in position 1799 may interchange the thymine and adenine nucleobases, thus engendering a replacement of valine ( v) by glutamate(E) at codon 600 (V 600 E) of the BRAF protein. The mutation may be detected in 80% to 90% of hairy cell leukaemia14 The BRAF V600E mutation may activate the BRAF gene via autophosphorylation of the protein with a consequent decline of the MEK-ERK signalling network, thereby ensuring amplification of genes inciting cellular multiplication and continuation2. Mutant BRAF V 600 E gene may not be elucidated in adjunctive B cell chronic lympho-proliferative conditions7, excluding occasional instances of chronic lymphocytic leukaemia or multiple myeloma. The BRAF mutation is a characteristic molecular feature , a contemporary diagnostic tool and a possible therapeutic approach of targeting BRAF by employing BRAF inhibitors15. A lack of BRAF genetic mutation may arise in 10% -20% of hairy cell leukaemia which comprises of a subcategory of inferior prognosis. The particular instances necessitate an demarcation from a patients with a probable mutation in exon 11(F468C, D449E)16. The BRAF V600E mutation may reappear in diverse solid tumefaction such as the cutaneous melanomas, pulmonary, ovarian, bladder, thyroid or prostate malignancies, cholangiocarcinoma and gastrointestinal stromal tumours/ sarcomas2.
Genetic Disease Progression
Refractory hairy cell leukaemia may delineate a repetitive neutralization of the cell cycle inhibitor CDKN1B / p27 in a minority (16 %) of the instances. Supplementary mutations of KLF2 genes may be exemplified in an estimated one third (30%) of marginal zone lymphoma ( MZL) and diffuse lymphomas( DLBCL). KLF2 is a transcription factor which regulates the maturation and differentiation of numerous B cell subpopulations, especially marginal zone B cells2. Along with BRAF mutations, the two frequent mutations discerned in hairy cell leukaemia may be the histone methyltransferase KMT2C ( MLL3) in 15% instances and the CDKN1 B mutation appearing in 11% individuals2 (Table 1, Table 2, Table 3, Table 4).
Table 1. Preliminary investigations for detecting hairy cell leukaemia(5).| Recommendations | Specialized procedures |
| Diagnosis and initial assessment | |
| Complete blood count | |
| Peripheral blood smear review | A Wright’s stain for white blood cell differential count to identify leukaemia cells |
| Immuno-phenotypic analysis by flow cytometry | Immune reactive for CD19+, CD20+, CD11c+,CD25+, CD103+,CD123+ CD200+ & immunoglobulin light chain restriction for circulating mononuclear cells. |
| Bone marrow aspiration and trephine biopsy | A haematoxylin and eosin stain, reticulin stain and immune reactivity for CD20+, annexin-1, DBA44 & VE-1( BRAF V600E), identification of the genetic or BRAF V600E mutation by allele specific PCR , a sequence analysis or an immune stain to confirm diagnosis & the degree of bone marrow infiltration |
| Complete history and physical examination | Including renal function tests for patients requiring nucleoside analogue therapy |
| Optional imaging studies | Chest X-ray to evaluate infection, CT or abdominal ultrasound to assess organomegaly and/or lymphadenopathy, particularly with patients on clinical trials or with concordant systemic symptoms. |
| Hepatitis serology for employing the anti CD20 monoclonal antibody | |
| Differential Diagnosis | Consider hairy cell leukaemia, hairy cell leukaemia variant, splenic marginal zone lymphoma, splenic diffuse red pulp small B cell lymphoma( with specific immune phenotype) |
| Indications for treatment | |
| Haematological parameters consistent with commencement of treatment | A minimal of one parameter : haemoglobin<11gm/dl, platelets<100,000/µL or absolute neutrophil count < 1000/µL. |
| Clinical features or systemic symptoms appropriate for therapy | Symptomatic organomegaly, progressive lymphocytosis, lymphadenopathy, unexplained weight loss(>10% body weight in the preceding six months), excessive fatigue( grade >2) |
| Specific Mutation | Percentage Alterations |
| MAPK pathway | |
| BRAF V600E | 70%-100% |
| MAPK 2K1 | 0.0%- 22% |
| Cell Cycle | |
| CDKN1B( p27) | 11%-16% |
| CCND3 | 0% |
| NOTCH pathway | |
| NOTCH 1 | 4%-13% |
| NOTCH 2 | 0.0% - 4% |
| Epigenetic regulators | |
| KMT2C( histone methyltransferase ) | 15% |
| ARID 1A( SWI/SNF family) | 4% |
| Transcription factors | |
| TTN | 4% |
| KLF2 | 13%-16% |
| NF ₭B pathway (MYD88,TNFAIP3),Spliceosome (U2 AF1,TP53),TF repressor (BCOR) | 0% |
| Preliminary Treatment |
| Cladarabine administered subcutaneously or as a continuous, intravenous infusion. |
| Pentostatin administered intravenously with a deliberation upon the renal function |
| Treatment at Relapse |
| Following confirmation of the preliminary diagnosis, a review of previously employed therapeutic protocols with the identification of patients with poor risk ( severe anaemia, spleen > 10 cm below the left costal margin, atypical immune phenotype, absence of BRAFV600E mutation) |
| Indications of repetitive therapy simulating the initial criterion for therapeutic commencement particularly symptomatic disease (splenomegaly) or progressive anaemia, thrombocytopenia or neutropenia. |
| If previous remission was > 24 months , a retreatment with a purine analogue with a concurrent anti CD 20 monoclonal antibody or a clinical trial |
| If previous remission was > 60 months, consider initiating the preliminary therapy. |
| If previous remission was < 24 months, alternative therapy with investigational agents( such as vemurafenib) may be employed following the ascertainment of a precise diagnosis. |
| Previously approved therapeutic modalities may be beneficial( such as interferon ᾳ, splenectomy ,rituximab) |
| Category of response | Approved criterion for response |
| Complete Remission(CR) | Near normalization of peripheral blood counts( haemoglobin > 11gm/dl without transfusion, platelets > 100,000/µL, absolute neutrophil count > 1500/µL. Regression of palpable splenomegaly, absence of morphological evidence of hairy cell leukaemia on peripheral smear and bone marrow. |
| Timing of response assessment | Response evaluation on the bone marrow treated with cladarabine should be done after 4-6 months following discontinuation of therapy. Patients on pentostatin may require a bone marrow evaluation after near normalization of the peripheral blood counts and regression of palpable splenomegaly. |
| Complete Remission with/ without Minimal Residual Disease(CR with/without MRD) | In patients who have achieved a CR, an immune-histochemical evaluation of the percentage of MRD will demarcate betwixt individuals on CR with or without evidence of MRD |
| Partial Response(PR) | A near normalization of the peripheral blood counts( as in CR) with a minimal 50% improvement of organomegaly and a bone marrow biopsy infiltrated with hairy cells. |
| Stable Disease(SD) | Patients who do not meet the criteria of objective remission following therapy may be considered to have SD. As the patients are treated for specific reasons such as constitutional symptoms or a decline in the haematological parameters, the category of stable disease may not be an acceptable response |
| Progressive Disease(PD) | Amplifying disease related systemic symptoms, augmented organomegaly by 25% or a 25% decline in the haematological parameters may qualify as PD. A distinction from reduced haematolo gical parameters due to therapeutic myelo-suppression may be mandated |
| Hairy Cell Leukaemia in relapse | Morphological relapse is defined as the reappearance of hairy cell leukaemia in the peripheral blood, the bone marrow biopsy or both by appropriate stains in the absence of a haematological relapse. Haematological relapse is defined as a reappearance of cytopenia(s) or parameters below the threshold as defined for complete response and partial response. Whereas no treatment is required for morphologic relapse , therapeutic decisions for a haematological relapse may be obtained by evaluating benchmarks such as haematological values necessitating intervention or reoccurrence of disease related symptoms. |