Abstract
Antibodies and antibody fragments, especially single-domain antibodies known as nanobodies, are important tools in diagnostics, research, and therapeutics. In a conventional antibody, light and heavy chains contribute to the formation of the antigen binding site. In addition to conventional antibodies, old and new world camels also have heavy-chain antibodies (hcAbs), which lack the light-chain antibodies that usually bind to the antigen, as well as single domain antibodies, the VHH domain, which are the smallest antigen-binding fragments and have high solubility, stability, and specificity.
A VHH library against E. coli lipopolysaccharide (LPS) was produced using the camel immune system. E. coli strains from dead camel calves were isolated to extract the LPS and used to immunize a 2-year-old female camel. After isolating mononuclear lymphocytes for RNA extraction and amplification of the VHH gene, the PCR product was cloned into the pF1AT7 Flexi vector and transformed into JM109 E. coli competent cells by heat shock, resulting in a comprehensive VHHs library with 6.9 × 104 cfu/µg. The VHHs were expressed and screened with ELISA and PCR. Eleven colonies were positive by PCR, six of which were sequenced and submitted to Genbank compared with GenBank data to confirm the production of nanobodies with a similarity >90%.
Author Contributions
Academic Editor: Mohammed A Elmetwally, Associate Professor of Theriogenogy.
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2021 Faysal Abdishakur Hassan, et al.
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
Authors Faysal Abdishakur Hassan and Abdelhaq Anouassi were employed by the company Advanced Scientific Group. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Citation:
Introduction
Antibodies are essential tools in diagnostics, basic research, and therapy. Immunoglobulins (Ig), conventional antibodies, are large Y-shaped glycoproteins 1 with a molecular weight of approximately 150 kDa. They are mainly synthesised in plasma cells as a first defence mechanism of the body and are also used in research experiments and medical applications. They are usually classified according to their heavy-chain isotypes into IgG, IgM, IgE, IgA, and IgD, which vary in both structure and function. These antibodies contain two heavy and two light chains; all four chains contribute to the mechanism of reaction. In addition to these conventional antibodies, camelids have three IgG isotypes: IgG1, IgG2, and IgG3. IgG1 has a typical conventional antibody structure whereas IgG2 and IgG3 are referred to as heavy-chain antibodies (HCAbs) because they lack light chains as well as CH1. Their nomenclature is based on the biophysical properties of IgG isotypes such as their separation by chromatography into three isotypes 2.
Unlike conventional antibodies, the antigen binding site of a heavy-chain antibody (camelid antibodies) can bind with antigens with a single domain heavy chain called a VHH domain 3. Overall, the single domain heavy chain antibodies of camels are the smallest antigen-binding fragment (~15 kDa) and have high stability, solubility, and specific antigen-binding capacity, which provides key defences against pathogenic organisms and toxins 2, 4. A significant amount of research has focused on the camelid antibody because it has been demonstrated to have excellent potential for applications in immunotherapy, diagnostics, and biosensors 2. The biosensor function is particularly useful for targeting cancer cells, inflammatory responses, autoimmune diseases, and viruses 5. These nanobodies can be easily manufactured in milligram quantities in E. coli and can be stored for extended periods 6, 7. This type of production would normally assist with the creation of a bank of nanobodies with numerous applications for cell biology research 8.
More than two decades have passed since the discovery of naturally occurring single-chain antibodies. They have been used as diagnostic tools in a number of studies 6, 7. A number of different nanobodies have been generated against many diverse individual antigens, such as tumour receptors 9, 10, HIV 11, clostridium botulinum neurotoxin, 12 and many other microbial antigens.
In camels, E. coli causes major neonatal diarrhoea resulting in up to 40% loss of camel calves, especially when they are not treated well during the first few days of their life 13. That is a significantly heavy loss in the camel breading industry. E. coli outbreak in our breeding centre resulted in high mortality in camel calves in 2013. In 2013, Advanced Scientific Group (ASG) lost approximately 30% of its newly born racing camels within their first 10 days of life (Anouassi et al., unpublished data). All post-mortem reports indicated that colisepticemia or colibacillosis was the cause of death. Colibacillosis and colisepticemia mostly occur in the first weeks of camel life (one to four weeks), causing high fever, yellowish watery diarrhoea, dehydration, and death. The aim of this study was to produce a single-domain antibody (sdAb) against the lipopolysaccharide (LPS) of E. coli strains isolated from dead calves for possibility of use in the future as passive immunisation for neonatal camels, especially during the first few days of their life.
The immune system of a dromedary camel was used to generate VHH libraries against E. coli LPS. The LPS of E. coli was chosen as an antigen because it is the major component of the outer membrane of all Gram-negative lipid bilayers. It contains lipid A, polysaccharide (O antigen) with repeating units of many lengths. Lipid A (endotoxin), the hydrophobic anchor of lipopolysaccharide (LPS), causes fever and septic shock in mammals. Overall LPS is a very powerful toxin which can activate the immune system of mammals at low concentration. The libraries were screened with specific VHH primers and sequenced. Additionally, ELISAs were performed for antigen-specific VHHs as well as PAGE electrophoreses to demonstrate the production of the nanobody.
Methods
Preparation of E. coli Lipopolysaccharide and Camel Immunization
The E. coli sample was isolated from the small intestines of a two weeks-old camel calve that showed E. coli septicaemia. The sample was cultured in lysogeny broth (LB) and incubated overnight at 37 °C. 1 mL of the culture suspension was placed in a 1.5 mL tube then centrifuged for approximately 30 s at 10,000g and then the supernatant was removed. E. coli LPS was then extracted using an LPS extraction kit (Intron biotechnology Korea) according to the manufacturer’s protocol.
A two-year-old female camel (Camelus dromedaries) was used in this experiment. To start the experiment, the camel was isolated together with 5 other camels for one week to allow the camel to overcome stress that may cause immunosuppression 14. Then, a 50 mL blood sample was collected before administering the animal with E. coli LPS (immunogen). The blood was allowed to clot for two hours at room temperature. The clotted blood was centrifuged for 5 min at 3,000 ×g. Then, the supernatant (serum) was stored at −20 °C and used as the ELISA negative control to compare the titre of the pre-immune serum and post-immune plasma when measuring the serum alteration induced by each antigen. Serum was made by four-fold serial dilutions.
The two-year-old camel was immunised six times with the freshly prepared LPS at two-week intervals. Each immunization included six 0.2 mL intradermal injections of E. coli LPS (1 mg/mL) 15. The LPS was gently mixed with an equal volume of complete Freund’s adjuvant (Santa Cruz Biotechnology, USA) to make an emulsion, then injected intradermally. The first, second, third, and fourth immunizations were administered in the base of the camel’s neck, near the bow lymph node. The fifth and sixth booster doses of LPS were emulsified with incomplete Freund’s adjuvant and administered in the pre-scapular region 16.
One week after completion of a 70-day immunization regime, 400 mL of peripheral blood was collected and mixed with sodium citrate as an anticoagulant, then immediately transferred to the lab for assessment.
ELISA Procedure
E. coli LPS was used as an antigen in enzyme-linked immunosorbent assay (ELISA) to monitor the immune response of the camel. Plate wells were coated with 100 µL of the E. coli LPS antigen at a concentration of 10 µg/mL in a coating buffer (50 mM sodium carbonate buffer, pH 9.6). The plate wells were covered and incubated for four hours at room temperature and then overnight at 4 °C. Wells were blocked with 1% bovine serum albumin (BSA) in phosphate buffered saline (PBS) (200 µL/well) and incubated with 100 µL of serum. After extensive washing, the bound camel antibodies were detected with a rabbit anti-camel IgG conjugated to horseradish peroxidase (HRP) (ABclonal China) and 100 µL of 1 mg/mL TMB was added to the reaction as a substrate. After colour developed in the solutions, 100 μL of 0.5 M sulphuric acid was added to each well and the microplate was read with a spectrophotometer at 450 nm.
Construction of a Library
Lymphocyte Separation, RNA Extraction and cDNA Synthesis.
For the construction of a library, peripheral blood mononuclear lymphocytes (PBMCs) were separated by (Lymphoprep™ solution) and total RNA was extracted using TRIZOL® Reagent according to the manufacturer’s protocol. The samples were then quantified by measuring the OD at 260 nm and cDNA was synthesised from 2.5 μg of RNA using the GoScript™ Reverse Transcription System (Promega, USA) to serve as a template for the first PCR reaction. The variable domains of heavy chains (VHs and VHHs) were amplified from the cDNA with two of one modified gene-specific primers, CALL001 and CALL002 17. CALL001: 5′-GTCCTGGCTGCTCTTCTACAAGG-3′; CALL002: 5′-GGTACGTG CTGTTGAACTGTTCC-3′; VHH-Rev: 5′- CAA ATT TG--GATGTGCAGCTGCAGGAGTCT GGRGGAGG-3′ (PmeI); VHH-For: 5′-CTAGTGCGGCCGCTGGAGACGGTGACCTGGGT--GCG ATC GCC ATG-3′ (SgfI) 14, 17. The nested primers with modification of the original sequencing and endonuclease restriction sites of SgfI and Pmel were introduced. All primers were made by Alpha DNA (Canada) and constituted in 10 mM Tris-HCl buffer (pH 8.5) to make 100 μM stock solutions. Eight PCR reactions of first PCR were performed, each reaction in a total volume of 25 µL of reaction mixture containing dNTPs mix (0.4 µM), CALL001 and CALL002 primers (0.4 mM each), GoTaq DNA polymerase (1 U) (Promega Madison, WI USA), PCR buffer supplied by the manufacturer, and 2 µL of first-strand cDNA reaction. The reaction was incubated at 95 °C for 5 min then 30 PCR cycles were performed, each cycle consisting of 45 s of denaturation at 94 °C, 45 s of annealing at 55 °C, and 45 s of extension at 72 °C. Then, the final extension step was performed for 10 min at 72 °C. All PCR reactions were pooled and 10 µL of the sample was used for 2% agarose gel analytics with 5 µL of 20.000X RedSafe nucleic acid staining solution (iNtRON Biotechnolgy, Korea) and electrophoresed at 90 V for 40 min in 1X TAE buffer (0.04 M Tris/acetate, EDTA 1 mM). A 100 bp ladder (Promega, Madison, USA) was used as the molecular weight marker. The PCR bands matching the VHHs bands, those lower than 500 bp, were cut out of the gel with a sterile scalpel. The DNA fragments from agarose were purified using an Agarose Gel DNA Extraction Kit (Roche Germany). The second PCR was amplified using modified VHH-Rev, and VHH-For primers (0.4 mM each) After obtaining a clear band of approximately 400 bp, the PCR products were purified using the QIAquick PCR Purification Kit (Qiagen) and DNA was eluted in the elution buffer. The concentration was measured by NanoDrop spectrophotometry. Restriction digests of the second PCR product and acceptor Flexi® vector were simultaneously performed using SgfI and PmeI digestion enzymes in a Flexi® vector system (Promega USA). Both the reactions were incubated at 37 °C for 30 min and then heated at 65 °C for 20 min. Then, the digested products were purified using a QIAquick PCR purification kit (Qiagen) and stored on ice. The acceptor Flexi® Vector and PCR product were incubated at room temperature for 1 h with a ligation enzyme according to the manufacturer’s protocol (Promega USA). Then, the products were transformed into high-efficiency E. coli competent cells JM109 (Promega); the selection of transformants was performed on LB plates supplemented with 100 µg/mL ampicillin suitable for the acceptor pF1AT7 Flexi® vector (Promega USA). The plates were cultured for 24 h and 48 h and the colonies were randomly selected and divided into two groups, 48 each, and sub-cultured overnight in LB broth in a 96 well plate. Plasmid DNA extraction was performed using a plasmid DNA extraction kit (Intron, Korea) and digested with SgfI and PmeI enzymes and PCR was performed using VHH primers. The PCR-amplified VHH products 6 were purified with QIAquick (QIAGEN, CA, USA) and directly sequenced with only a forward primer pair. Sequencing was performed with Bigdye Terminators on an ABI 3730 genetic analyser (Applied Biosystems, CA, USA) according to the manufacturer’s protocol.
Protein Expression
Each colony was transferred to 10 mL of LB media and incubated at 37 °C (with shaking) for 3-5 h. Once the culture reached 0.6 OD, 1 mL of culture was taken and centrifuged at max speed for 5 min in a 1.5 mL tube. The supernatant was discarded and the culture was expanded by adding 10 mL of LB with ampicillin antibiotic (room temperature). Then, the culture was incubated for 3 h at 37 °C (with shaking). The culture was induced by adding isopropyl β-D-thiopgalactopyranoside (IPTG; Sigma, Germany) to a final concentration of 0.5 mM and kept for 4 h at 37 °C with shaking. The mixture was centrifuged at 3,500 ×g for 20 min. The supernatant was discarded, the cells were re-suspended in ice-cold PBS, and the sample was re-centrifuged at 3,500 ×g for 20 min. Then, proteins were extracted using the Smart Bacterial Protein extraction kit (Intron Korea). 15% polyacrylamide SDS gel was prepared using Intron polyacrylamide. Then, 18 µL of bacterial protein extract was added to 6 µL of 4x SDS-PAGE loading buffer. The samples were boiled with a low molecular weight marker for 10 min at 95 °C and spun down at 3000 g for 5 min. Then, 15 µL of each sample was loaded into the SDS-PAGE and run at 90 V for 15 min and 200 V for 40 min. The gel was stained with SenSafe™ Fast Protein Staining Solution (Intron, Korea)
Results and Discussion
A healthy two-year-old dromedary camel was immunized with highly pure and immunogenic E. coli LPS antigen isolated from a dead neonatal camel. Subsequently, the VHH genes were amplified from mononuclear lymphocyte cDNA, encoding the variable domains of the heavy chain antibodies. First, the CALL001 and CALL002 PCR primer pair was used to obtain the specific product of VHH-CH2 700 bp exons. Then, VHH encoding gene fragments were amplified in the second PCR, which was purified from an agarose gel after cutting the 700 bp band obtained from the first PCR using VHH primer pairs. A 400 bp DNA fragment was generated (Figure 1), transforming the ligation of the VHH genes amplified with nested PCR and pF1AT7 Flexi vector (Promega USA) into JM109 E. coli competent cells (Promega USA.), and then the cells were plated on LB plates supplemented with 100 µg/mL ampicillin. A VHH library was generated after overnight incubation; successful growth was observed the next day and 48 independent clones that overexpressed the resistance gene on the LB agar plate were randomly picked and plasmid extraction was performed; 11 of them were PCR positive and clear bands of 400 bp fragments were evident (Figure 2), which revealed a correct insertion. The transformation efficiency (TE) of the library was 6.9 × 104 cfu/µg. To increase the VHH library size, the absolute amount of ligated DNA and the number of successful transformations are very important factors that must be considered. The cloning of the PCR amplicon with the corresponding restriction enzymes resulted in an open reading frame of the pF1AT7 flexi vector to the putative VHH domain, which facilitated transportation of the expressed protein to the bacterial periplasm to ensure the correct formation of essential disulphide bonds.
Figure 1. CALL primers (1 and 2) show double bands of 700 bp and 400 bp whereas the VHH primers (3,4 and 5) give only one band of 400 bp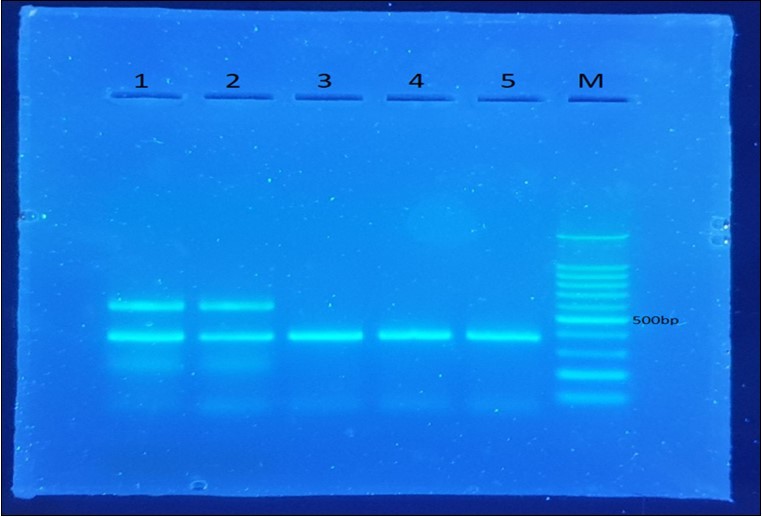
Figure 2. PCR of the VHH gene from plasmid DNA of transformation. 11 of them are indicated as PCR positive.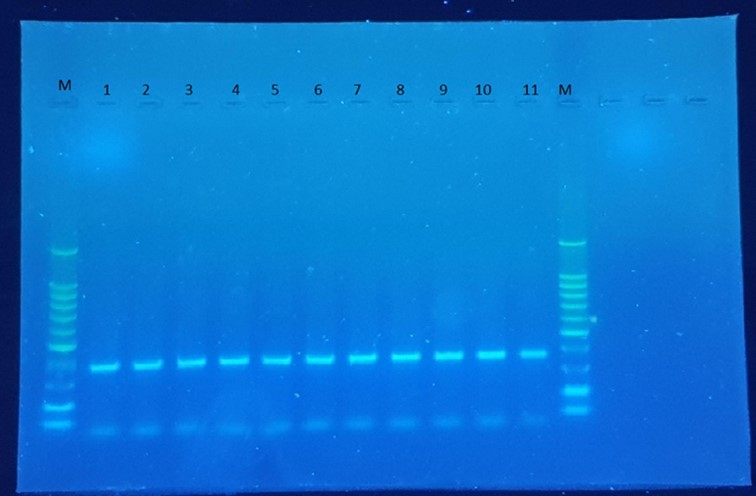
To select antigen-specific VHHs, mononuclear lymphocyte cells from immunised camels were used as a foundation to generate a comprehensive VHH library, because they should specifically contain mRNA encoding or LPS antigen-specific VHHs. This is important to ensure a large and diverse library and to maximise the mRNA yield. To do this, a large number of lymphocytes from peripheral blood is required. In brief, 300–400 mL of heparinised blood from the immunised camel was used. Subsequently, total RNA was extracted from the isolated mononuclear lymphocytes with a concentration of 2499 μg/mL, which was sufficient to perform cDNA synthesis using oligo dT primers in a GoScript™ Reverse Transcription System (Promega, USA).
To obtain a large VHH library, the ligation steps of the VHH amplicon and the corresponding pF1AT7 flexi vector as well as next transformation steps are very important. First, the VHH encoding gene fragment and the vector were digested by SgfI and PmeI, two rare-cutting restriction endonucleases, followed by ligation and transformed by heat shock into JM109 E. coli competent cells (Promega USA).
Six out of the eleven PCR-amplified VHH products from plasmid extraction were purified with QIAquick (QIAGEN, CA, USA) and directly sequenced with only a forward primer pair and submitted to Genbank using BankIt online software for NCBI and obtained accession numbers for six nucleotide sequences: BankIt2366596 seq1 MT792357 seq2 MT792358 seq3 MT792359 seq4 MT792360 seq5 MT792361 seq6 MT792362 and then compared and aligned with GenBank sequencing data. Amplicons (400 bp) were used as a query for searching for a homologous sequence in GenBank using Nucleotide BLAST (Basic Local Alignment Search Tool) in the GenBank database 18. All six anti-LPS VHH were sequences and showed a similarity of more than 95%. Colonies 1 and 2 had unique amino acid sequencing and they were classified into 5 families according to the distinct amino acid sequences in the CDR3 region, as shown in Figure 3. Simultaneously, the blast results comparing many immunoglobin heavy-chain sequences from dromedary camels, lamas, and vicunas showed a similarity of approximately 90% and no sequence matched more than 91% in the GenBank database (Figure S1, Supporting Information).
Figure 3. Protein sequencing alignments results of six colonies by ClustalW software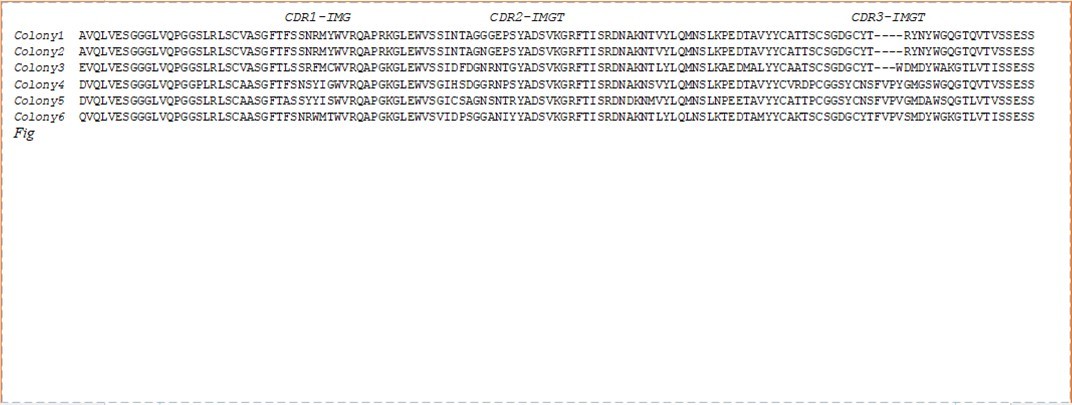
Figure S1. GenBank data showing similarity about 90% with first clone
The VHH sequence alignments of single clones are illustrated in Figure S2 (Supporting Information). To select a specific VHHs E. coli LPS antigen to evaluate the success of the process, ELISA measurement was used as a semi-quantitative assessment to verify this result and exclude unspecific VHHs. The pre-immunised serum was tested in parallel with a negative control. Eleven VHHs were selected on positive clones in PCR for ELISA and 9 of them were positive after the growth on LB broth medium with IPTG to a final concentration of 0.5 mM. Six of them are shown in Figure S3 (Supporting Information). However, nine sub-cultured colonies were protein-extracted and ELISA was performed using plates coated with antigen E. coli LPS. Colour was developed, which demonstrated that these nanobodies recognised their specific antigens screened for their ability to react with fixed antigens on the ELISA plate. To determine the size of the VHH libraries, E. coli cells were transformed with the flexi vector library and plated in serial dilutions, from which the colony forming units (cfu) were counted and calculated using the TE equation. The library size was 6.9 × 104 cfu/µg. LPS-specific VHHs were classified into different families derived from different amino acid sequences, especially in CDR3 hypervariable areas. The plasmids of the different families were extracted and digested with SgfI and PmeI endonucleases, as shown in Figure 4. Furthermore, positive PCR results by SDS-PAGE confirmed the production of nanobodies. The bands of the nanobodies were revealed by 15% SDS-PAGE at approximately 17 kD.
Figure S2a. Clustal O multiple nucleotide sequencing alignments
Figure S2b. Multiple protein sequencing alignments.
Figure S3. Result of ELISA test for VHH production of six colonies using two different antibodies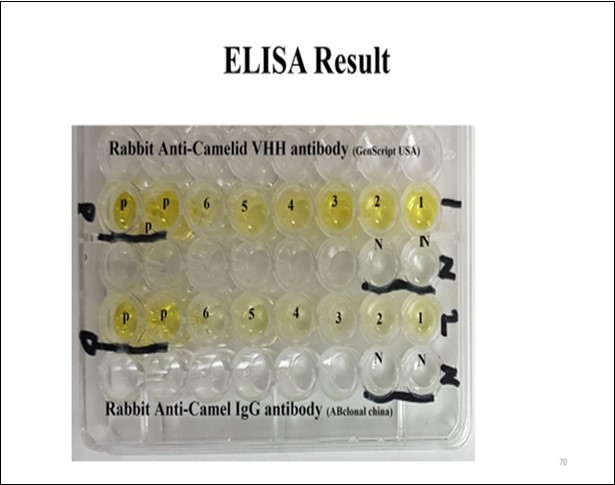
Figure 4. Plasmid extraction and vector insert digestion.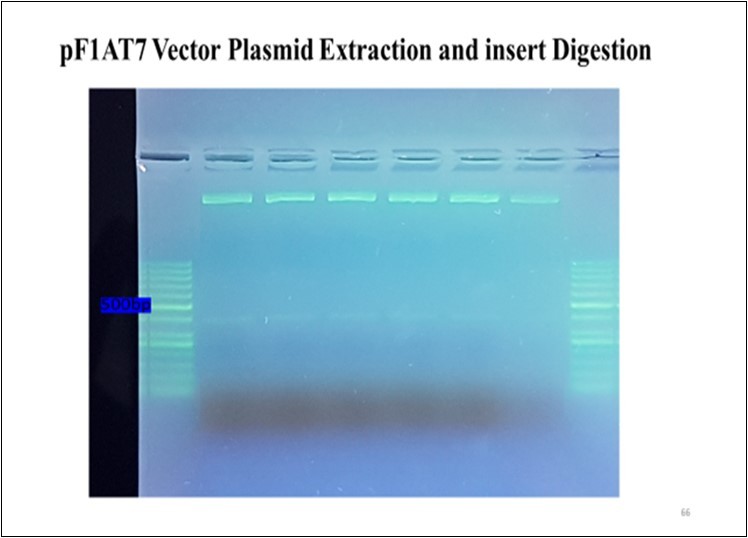
To produce nanobodies specific to LPSs of the E. coli cell wall antigens, the expression cloning vector Flexi® Vector was chosen (Promega, USA), with a protein-coding sequence flanked by SgfI and PmeI sites. These two rare-cutting restriction endonucleases helped to clone the VHHs gene acquired from immunised camels after amplifying with a previously tailed VHH primer pair with a recognition site for SgfI and PmeI endonucleases. The SgfI site is upstream of the start codon of the protein coding region, which induces the expression of a protein by reading the SgfI site, whereas the PmeI site comprises the stop codon for the protein-coding region and attaches a single valine residue to the protein.
Anti E. coli LPS nanobodies were produced from the expression cloning vector based on positive ELISA results, which led to recognition of nine colonies of expression nanobodies bound to the LPS antigen that was coated on the plate. However, eleven colonies were found to be positive for VHH primers via PCR on extracted plasmid DNA, including six that approximately matched with the positive ELISA tests. From all the PCR products, these six colonies were sequenced using only the forward primer. Positive PCR results were obtained through SDS-PAGE to confirm the production of the nanobodies.
E. coli causes diarrhoea via six different mechanisms, each with different virulence and pathotype results including septic infection, acute enteritis, haemorrhagic colitis, and diarrhoea. These pathotypes include enteropathogenic E. coli (EPEC), terohaemorrhagic E. coli (EHEC), enterotoxigenic E. coli (ETEC), enteroaggregative E. coli (EAEC), enteroinvasive E. coli (EIEC), and diffusely adherent E. coli (DAEC)
However, in our recent study ,19 we showed that none of the six pathotypes of E. coli were the cause of the death of neonatal camels. Although pathogenic E. coli strains cause infection in some animals, they can be commensal flora for others. Because the immune system activation in new born camels is weak, specifically in the first days of their life, infections with opportunistic microbial flora can result from ingestion of contaminated mother’s milk or unhygienic food where the host defence mechanisms are weak, particularly our finding was demonstrated that all E. coli isolates that killed neonatal camels were non-pathogenic. However, in this study we concluded that there is a possibility that other virulence factors are responsible for these calf mortalities and the nanobody from the the same animal can be used to defend themselves.
The production of nanobodies against E. coli to use as passive immunization for camel calves is very important, especially during the first week of their life, because they have no antibodies to fight disease without an adequate supply of mother’s colostrum as a source of nutrients and they are very susceptible to infection from the surrounding environment. Pathogenic E. coli strain K99 20 is widely used for passive immunization in cattle, horse, and camel farms, specifically in their first week of life.
In conclusion, passive immunization using the E. coli LPS nanobodies prepared in this paper could prevent a lot of camel calf deaths. Good hygiene is also essential and it is extremely important to maintain a clean environment.