Abstract
Ciliary neurotrophic factor (CNTF) is a well-tested, neuroprotective agent that has been shown to retard photoreceptor degeneration in several animal models of retinitis pigmentosa. The molecular mechanisms underlying CNTF-mediated neuroprotection are currently not understood. CNTF could act directly on photoreceptors or it could act indirectly by stimulating Müller glial cells to produce photoreceptor neuroprotective agents. To better characterize CNTF action on Müller cells, we have studied signaling pathways activated by CNTF using an established retinal Müller cell line, rMC-1. RNA was isolated from CNTF-treated cultures, and suppressor of signal transducer and activator of transcription (SOCS3) and Glial fibrillary acidic protein (GFAP) transcript levels were assessed by quantitative real-time PCR. Immunoblotting was used to examine activation ofmitogen activated protein kinase (ERK1/2/MAPK) and phosphoinositide 3-kinase (PI3-K)/Aktpathways in response to CNTF. Additionally, the level of5' AMP-activated protein kinase (AMPK), an enzyme that plays a key role in cellular energy homeostasis levels, was determined by immunoblotting. CNTF treatment resulted strong upregulation of SOCS3 and GFAP transcripts that were blocked by expression of a dominant-negative STAT3 mutant. CNTF treatment also resulted in transient activation of ERK1/2/MAPK but not PI3K/Akt signaling pathway. There was no change in activation of AMPK. We conclude that CNTF treatment leads to stimulation of JAK-STAT and MAPK signaling pathways but not the PI3K/AKT pathway, associated with cell death, in Müller cells.
Author Contributions
Academic Editor: Federico Gonzalez-Fernandez, State University of New York Buffalo
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2016 Vijay P. Sarthy, et al.
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Introduction
Ciliary neurotrophic factor (CNTF) is a neuroprotective agent that has been shown to retard rod photoreceptor degeneration in genetic models of retinitis pigmentosa or in animals with light damage, and is currently in clinical trials for Retinitis pigmentosa and Age-related macular degeneration 1. Its mode of action is not established although it has been postulated that CNTF could act indirectly by stimulating Müller glial cells to release factors that protect photoreceptors 1, 2, 3, 4, 5. To characterize growth factors and cytokines induced in Müller cells, we recently carried out microarray analysis of GFP+-Müller cells that had been flow-sorted from eyes injected with CNTF 6. We found that CNTF treatment leads to robust transcriptional activation of several genes in Müller cells in situ. One or more of the gene products such as Endothelin 2 and Transforming growth factor β1, might be responsible for the neuroprotective action of CNTF.
CNTF belongs to the Interleukin-6 (IL-6) family of cytokines, which includes CNTF, leukemia inhibitory factor (LIF), oncostatin M, cordiotrophin-1, IL-6 and IL-11 7. These cytokines share one or more of the receptor subunits, gp130 and LIFRβ, and signal through the Janus kinase-signal transducer and activator of transcription (JAK-STAT) signaling pathway 8. Activation by CNTF requires the CNTF receptor α (CNTFα), and CNTFα, LIFRβ and gp130 are widely expressed by neurons and glia in the CNS 7. CNTF acts on cells primarily by stimulating the JAK-STAT pathway 8. Additionally, CNTF appears to stimulate cell survival, through activation of mitogen activated protein kinase (MAPK) (also known as extracellular signal-regulated kinase (ERK) kinase) and phosphoinositide 3-kinase (PI3-K)/Akt pathways 1, 9. To investigate signaling pathways activated by CNTF in Müller cells, we have studied the effect of CNTF on activation of JAK-STAT, ERK1/2/MAPK and AKT pathways. In the present study, we have employed an established Müller cell line (rMC-1) to directly study CNTF signaling in Müller cells 10, thereby avoiding potential contribution from neighboring retinal neurons or other glial cell types
Intraocular CNTF injection has been shown to result in activation of the JAK-STAT pathway in rodent Müller cells 2, 3. In a previous study, we also reported that CNTF treatment of rMC1 results in a 4 to-5-fold increase in phosphor-STAT3 level, indicating activation of the JAK-STAT pathway in rMC-1 cultures 11. To further assess involvement of this signaling pathway, we have studied the effect of CNTF on suppressor of signal transducer and activator of transcription 3 (SOCS3) induction in rMC-1 cultures.
SOCS3 has been shown to be the dominant negative regulator of JAK-STAT in multiple cell types 13. Rapid and efficient termination of CNTF action is essential for controlling STAT activity as well as to prevent potential side effects of prolonged cytokine exposure, and SOCS3 appears to be a major player in this process. To investigate whether CNTF action is under the control of SOCS3 in Müller cells, we examined SOCS3 mRNA levels in rMC-1 cultures treated with CNTF.
Retinal Müller cell line 1 (rMC-1) grown in Dulbecco’s Modified Eagle Medium (DMEM) containing 10% Fetal Bovine Serum were switched to serum-free medium for a minimum of three hours prior to treatment with CNTF (R&D Systems, Minneapolis, MN) at 50 ng/ml for 0, 15, 30, 60 and 90 min. Total RNA was prepared from control and CNTF-treated rMC-1 cultures, and. RealTime quantitative polymerase chain reaction (RT-qPCR) was carried out using oligonucleotide primers against either Socs3 or Gfap.
Within 15 min after CNTF treatment, there was a 2.5 fold increase in SOCS3 mRNA, and by 30 min there was maximal (3.5-fold) increase in SOCS3 mRNA (Figure 1A). This response was transient and SOCS3 mRNA level decreased back to baseline by 90 min after CNTF treatment.
In order to test whether SOCS3 upregulation after CNTF treatment involves activation of the JAK/STAT pathway acting through tyrosine phosphorylation of STAT3, rMC-1 cultures were transfected with a dominant negative STAT3 plasmid, pCAGGS-neo-HA-Stat3F (dnSTAT3) using lipofectamine (LifeTechnologies Inc). This mutant plasmid disrupts the normal activation of the JAK/STAT pathway by binding to STAT3 and preventing proper dimerization of the phosphorylated STAT3 14. After transfection with dnSTAT3 mutant followed by CNTF treatment, there was no increase in SOCS3 mRNA in rMC-1 cultures (Figure 1B). This result proves that transcriptional activation of SOCS3 is mediated by STAT3 complex in the JAK-STAT pathway.
In glial cells, there is good evidence that CNTF treatment leads to induction of the Glial fibrillary acidic protein (GFAP), a hallmark of gliosis in the nervous system 15, 16. In agreement with this data, CNTF treatment leads to increased GFAP mRNA in rMC-1 cultures (Figure 1C). Within 15 min after CNTF treatment, there was a 10-fold increase of GFAP mRNA, and by 60 min there was maximal (30-fold) increase in GFAP mRNA (Figure 1). Surprisingly, the CNTF response was transient and GFAP mRNA level decreased back to baseline by 90 min after CNTF treatment (Figure 1). The increase in GFAP mRNA level was suppressed by transfection with the dominant-negative STAT3 mutant prior to treatment with CNTF showing that GFAP gene activation is mediated through phosphorylation of STAT3. GFAP mRNA level was reduced from 30-fold in non-transfected cells to less than 5-fold in transfected cells (Figure 1).
Figure 1. SOCS3 and GFAP transcripts are induced by CNTF treatment. (A) An increase in SOCS3 mRNA level in rMC-1 Müller cells was seen in response to treatment with CNTF, with a peak at 30min. (B) rMC-1 cells transfected with a dominant negative STAT3 mutant prior to stimulation with CNTF, results in no change in SOCS3 mRNA level in response to CNTF. GFAP is induced by CNTF treatment. (C) Increase in GFAP mRNA in rMC-1 Müller cells at 15, 30, 60, and 90 min in response to treatment with CNTF. (D) rMC-1 cells transfected with a dominant negative STAT3, mutant prior to stimulation with CNTF, show little GFAP mRNA induction in response to CNTF. The PCR primers used were: Socs3 (Forward) 5’-TACCC-TCCAGCATCTTTGTCGGAA-3', (Reverse) 5'-ATACTGGTCCAGGAACTCCCGAAT3'; Gfap (Forward) 5'-GGAAATTGCTGGAGGGCGAAGAAA-3', (Reverse) 5'-TGTGAGCCTGTATTGGGACAACT-3'; Gapdh (Forward) 5'-TGTGATGGG-TGTGAACCACGAGAA-3' and (Reverse) 5'-GAGCCCTTGCACAATGCCAAAGTT-3'.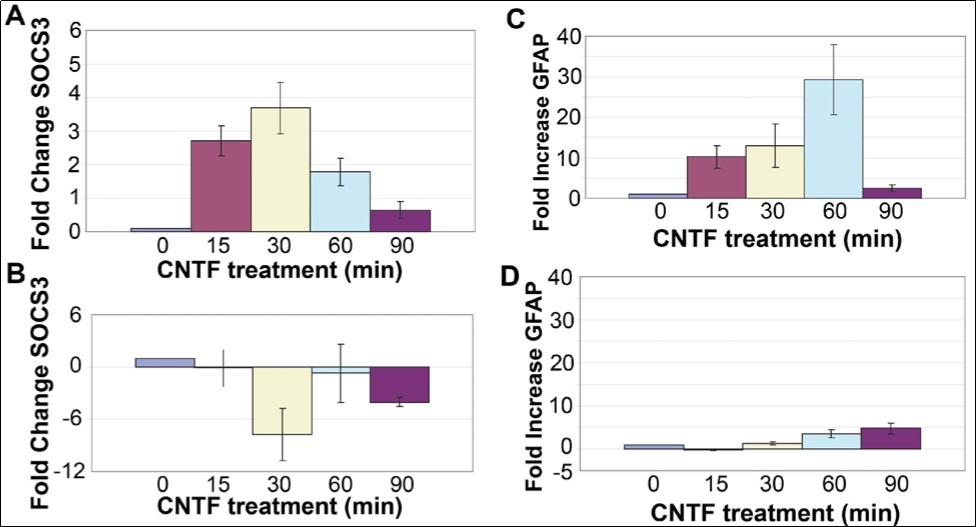
In agreement with previous reports, our studies showed that CNTF treatment leads to rapid activation of JAK-STAT pathway in Müller cells 2, 3. As activated JAKs are known to mediate recruitment of other molecules such as MAP kinase and PI3K/Akt 9, we also examined whether CNTF treatment resulted in activation of AKT and ERK1/2/MAPK pathways in Müller cells.
rMC-1 cultures were treated with CNTF for times from 15 to 90 min, and proteins were extracted from the cultures and Western blotted with antibodies against p44/42 MAPK (Erk1/2), Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204), Akt and Phospho-Akt (Ser473). All the antibodies (polyclonal) were ordered from Cell Signaling Technology, and were made in rabbits. Densitometry of the blots showed that CNTF treatment did not change the levels of pAKT upto 90 min ((Figure 2). In contrast, there was a 5.2 ±1.1-fold (n=3) increase in P-MAPK level by 15 min, and a substantial decrease to 1.9 ± 0.42-fold (n=3) by 30 min with P-MAPK level returning to baseline by 60 min (Figure 2B).
CNTF has been proposed to promote photoreceptor survival by reducing the metabolic stress in photoreceptors 1. Therefore, we examined whether CNTF treatment leads to activation of the 5' AMP-activated protein kinase (AMPK), a threonine/serine kinase that plays a key role in cellular energy homeostasis 17. In our experiments, CNTF treatment did not change AMPK levels in rMC-1 cultures (Figure 2).Taken together, our studies show that CNTF treatment leads to rapid activation of JAK-STAT and MAPK signaling pathways in Müller cells. This is not surprising as components of activated JAK-STAT pathway promote activation of other signaling pathways such as the ERK1/2/MAPK pathway that result in the recruitment of additional transcription factors that could lead to the synthesis and secretion of photoreceptor neuroprotective agents by the activated Müller cells or neighboring Müller cells.
Figure 2. Activation of AKT, MAPK and AMPK pathways in Müller cells. (A). Treatment of rMC-1 Müller cells with CNTF did not significantly alter the levels of P-AKT. (B) Treatment of rMC-1 with CNTF led to a sharp increase in the level of P-MAPK at 15 min. The level decreased substantially by 30 min and returned to baseline by 60 min. (C) CNTF treatment did not alter P-AMPK level.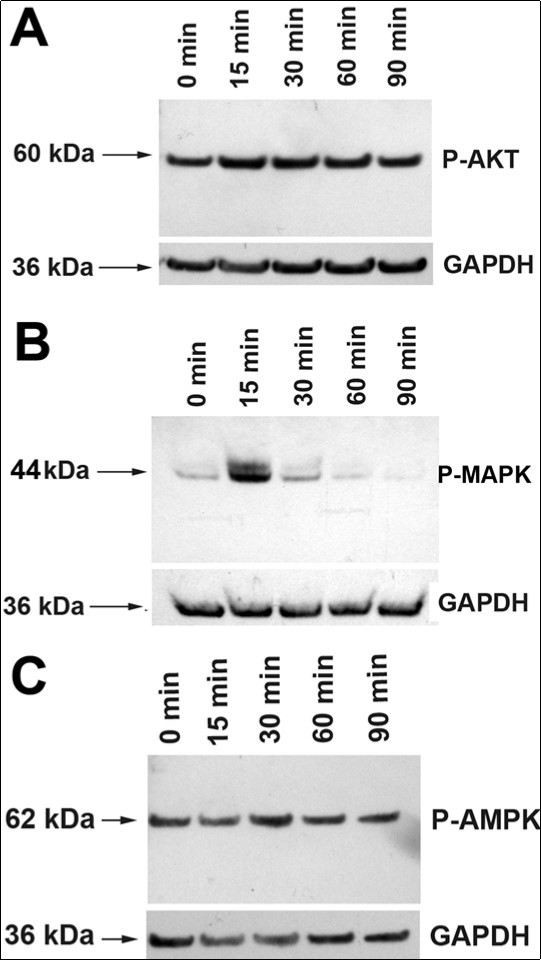
An important feature of the present study is that it examined the direct action of CNTF on Müller cells, and differs from in vivo studies that are limited by potential contribution from factors released by neurons or other glial cell types. As our experiments employed a Müller cell line, whether Müller cells in vivo behave in the same manner remains unknown. Nonetheless, our experimental findings are in excellent agreement with a previous study, which reported that intraocular injection of LIF, a close relative of CNTF, results in rapid activation of JAK-STAT and ERK1/2 (MAPK) pathways but not the PI3K/Akt pathway in the mouse retina 18. One notable difference is that LIF treatment in vivo leads to persistent activation of the JAK-STAT pathway but a transient activation of the ERK1/2 (MAPK) pathway where as in vitro CNTF treatment results in transient activation of both JAK-STAT and MAPK pathways. Whether this disparity in cytokine response is due to differences in the cytokines or in vivo vs in vitro effects remains to be determined.
Acknowledgements
We thank Dr. Toshio Hirano, Osaka University, Japan for kindly providing us with the plasmid, pCAGGS-neo-HA-Stat3F (dnSTAT3). This work was supported by NIH grant, EY019325 and by unrestricted funds from Research to Prevent Blindness, Inc.