Abstract
Renal fibrosis was a chronic and progressive process affecting kidneys in chronic kidney disease (CKD), regardless of cause. Although no effective targeted therapy yet existed to retard renal fibrosis, a number of important recent advances have highlighted the cellular and molecular mechanisms underlying the renal fibrosis. The advances including TGF-β/Smad pathway, oxidative stress and inflammation, hypoxia and gut microbiota-derived from uremic solutes were highlighted that could provide therapeutic targets. New therapeutic targets and strategies that are particularly promising for development of new treatments for patients with CKD were also highlighted.
Author Contributions
Academic Editor: Ying-Yong Zhao, Northwest University, China.
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2018 Shi-Xing Ma, et al.
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Introduction
Chronic kidney disease (CKD) had a high prevalence all over the world and was closely associated with high mortality 1, 2, 3. The prevalence of CKD was estimated to be 8-16% worldwide. In patients over 64 year old, the prevalence elevated to 23.4-35.8%, indicating increasing age contributed to elevate CKD. The yearly economic costs of medicine care for patients with CKD and or end-stage renal disease over age 65 were $60 billion, representing 24% of total Medicare expenditures in 2011 in America. According to the Kidney Disease Outcomes Quality Initiative, the international guidelines define and classify CKD as decreased renal function shown by glomerular filtration rate (GFR) of less than 60 mL/min per 1.73 m², or markers of kidney damage, or both, of at least three months duration, regardless of underlying cause 1. CKD were divided into five stages as follow: Stage 1: Kidney damage (pathological abnormalities or markers of damage including abnormalities in blood or urine tests or in imaging studies) with normal or raised glomerular filtration rate (≥90 mL per min per 1.73 m2); Stage 2: Glomerular filtration rate 60–89 mL per min per 1.73 m2 with evidence of kidney damage; Stage 3: Glomerular filtration rate 30–59 mL per min per 1.73 m2; Stage 4: Glomerular filtration rate 15–29 mL per min per 1.73 m2; Stage 5: End-stage renal failure; glomerular filtration rate <15 mL per min per 1.73 m21, 4, 5.
Renal fibrosis was characterized as a common endpoint of diverse CKD which resulted in functional damage ultimately leading to terminal renal failure 6, 7, 8, 9. Renal fibrosis is generally regarded as the dark side of tissue repair mechanisms. Fibrogenesis might be involved in the tubulointerstitium resulting in tubulointerstitial fibrosis, glomeruli resulting in glomerulosclerosis or the arterial vasculature resulting in atherosclerotic lesions 5, 10. Various action mechanisms were implicated in renal diseases and renal fibrosis 11, 12, 13, 14, 15, 16, 17, 18. Knowledge of the complex pathophysiological mechanisms contributed to CKD remains limited. In this review, we verify the critical roles of transforming growth factor-β (TGF-β)/Smad pathway, oxidative stress and inflammation, hypoxia and gut microbiota-derived from uremic solutes in the pathophysiology of CKD and renal fibrosis, summarize the action mechanisms of renal fibrosis, and discuss the effects of these mediators in the context of renal fibrosis.
TGF-β/Smad in Renal Fibrosis
TGF-β was essential for normal tissue development, repair and maintenance for organ functions. TGF-β1 was known as an antiinflammation cytokine 18. It produced anti-inflammatory effects through inhibition of mitogenesis and cytokine responses in glomerular cells and inhibiting infiltrating cells 18. Transforming growth factor-β (TGF-β1) knockout mice showed multi-organ inflammation and TGF-β1 deficient mice exhibited lethal inflammation and die within three weeks 19. Similarly, deletion of TGF-β1 or transforming growth factor-β receptor type II (TGFβRII) genes has been shown to cause autoimmune diseases 20, 21. Mice over-expressing latent TGF-β1 were protected against inflammation and renal fibrosis in obstructive nephropathy and glomerulonephritis 21, 22, 23. Although TGF-β-induced inhibition of NF-κB-mediated renal inflammation through induction of Smad7-dependent IκBα has been recently demonstrated 21, 23, the signaling action mechanisms of its anti-inflammatory action remain unclear. Yet, upregulated TGF-β1 was related to pathological disorders in many kidney diseases 24, 25.
There is extensive evidence pointing to TGF-β1 upregulation and its role in the pathogenesis of renal fibrosis in both animal models and patients with CKD 18, 26. TGF-β1 mediated progressive renal fibrosis by stimulating production and suppressing degradation of extracellular matrix (ECM). Moreover, TGF-β1 caused renal fibrosis by the transformation of tubular epithelial cells to myofibroblasts through epithelial-to-mesenchymal transition (EMT) 23. The central role of TGF-β1 on EMT and renal fibrosis has been confirmed by many experiments which indicated the ability of TGF-β1 blockade with decorin, neutralizing TGF-β antibody or anti-sense oligonucleotides to attenuate renal fibrosis 18. Direct evidence for the causal role of TGF-β1 in renal fibrosis is confirmed in mice over-expressing an active TGF-β1 form 27. TGF-β has been shown to serve a critical mediator in the pathogenesis of glomerulosclerosis in patients with glomerular diseases, such as lupus nephritis, immunoglobulin A nephropathy, membranous nephropathy, focal and segmental glomerulosclerosis and diabetic nephropathy. The upregulation of the three TGF-β isoforms and TGFβRI and TGFβRII has been uncovered in the glomeruli and tubulointerstitium in kidney diseases 28. Upregulation of TGF-β1 caused excessive ECM productions, reduced ECM-degrading proteinase activity and upregulated proteinase inhibitor, that resulted in excessive ECM deposition. In progressive podocyte-associated glomerular diseases, excessive TGF-β1 expression in the podocytes has been indicated the role of TGF-β1 in podocyte injury in patients with IgA nephropathy, focal and segmental glomerulosclerosis (FSGS) and diabetic nephropathy 29. Tubular and glomerular TGF-β expression was increased in early and late stages of diabetic nephropathy and inversely correlates with glycemic control in diabetic patients 30. TGF-β1 expression was stimulated by glomerular stretch and hyperglycemia in early stage, and by angiotensin II, advanced glycation end-product and platelet-derived growth factor 30. Angiotensin II has been demonstrated to raise expression of TGF-β1 and its receptors 31, 32.
Mounting studies have identified Smad2/3 as two major downstream mediators of the actions of TGF-β1 (Figure 1). In the context of renal fibrosis, Smad2/3 are activated in both patients and animal models with CKD of diverse etiologies such as hypertensive nephropathy 31, 33, 34, obstructive kidney disease 35, remnant kidney disease 36, 37, chronic renal allograft injury 38, diabetic nephropathy 39, 40, 41 and drug-induced nephropathy 42. Many fibrogenic genes including plasminogen activator inhibitor-1, tissue inhibitor of metalloproteinase-1, connective tissue growth factor, proteoglycans, integrins and collagens have been shown to be the downstream targets of TGF-β/Smad3 signaling 43. These observations demonstrate the central role of Smad3 in TGF-β/Smad signaling-mediated renal fibrosis.
Figure 1. The molecular mechanisms of renal fibrosis. Once an injury occurs in kidney, activated NF-κB and TGF-β/Smad pathways, as well as inhibited Nrf2 pathway were observed. The activation of NF-κB and TGF-β/Smad pathway induce inflammation and fibrosis, while the activation of Nrf2 pathway results in anti-inflammatory effects.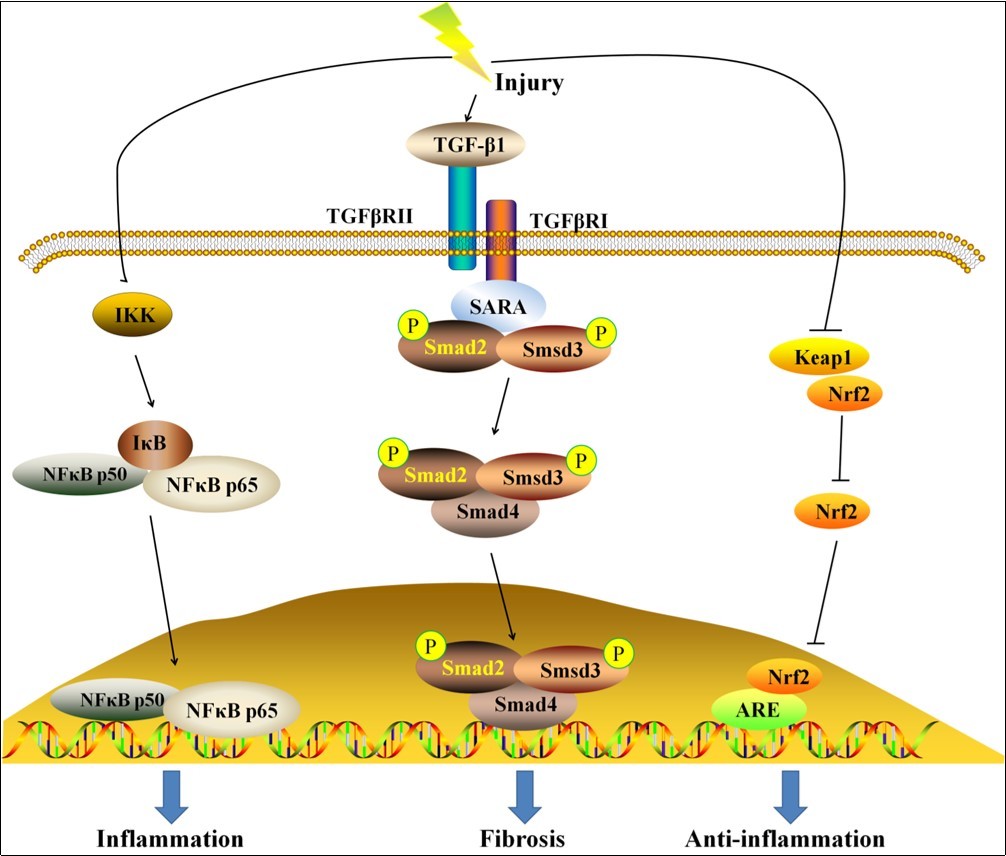
Many natural products have been widely used as anti-fibrotic agents 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56. Poricoic acid ZC, Poricoic acid ZD and poricoic acid ZE, isolated from the surface layer of Poria cocos, exhibited a strong inhibitory effect on renal fibrosis and podocyte injury. The findings showed that new RAS inhibitors poricoic acid ZC, poricoic acid ZD and poricoic acid ZE treatment significantly attenuated EMT production by inhibiting Wnt/β-catenin pathway activation and specific Smad3 phosphorylation by blocking the interaction of TGFβRI with Smad3 signaling in both TGF-β1- and angiotensin II-treated HK-2 cells as well as unilateral ureteral obstruction (UUO) mice 57. Similarly, renal fibrosis in a variety of animal models were mitigated via TGF-β/Smad pathways by administration of natural products, such as GQ5 58, curcumin 59, 60, arctigenin 61, resveratrol 62, sinomenine 63, berberine 64, 65, leonurine 66, rutin 67, bergenin 68, oxymatrine 69, 70, oleanolic acid 71, tanshinone IIA 72, astragaloside IV 73, 74, (+/-)-sinensilactam A 75 and epigallocatechin-3-gallate 76.
Oxidative Stress and Inflammation and Renal Fibrosis
Oxidative stress and inflammation played a central part in the pathogenesis and progression of CKD 77, 78, 79, 80, 81, 82. Renal fibrosis was a relatively common cause of CKD in humans. Rats or mice fed an adenine-containing diet exhibited severe renal fibrosis resembling that seen in humans 83. The renal fibrosis in this model was mediated by the renal tubular precipitation of dihydroxyadenine resulting in interstitial inflammatory cell infiltration, tubular epithelial cell injury, fibrosis and progressive deterioration of kidney function 84. Progressive renal disease was largely driven by inflammation and oxidative stress. Oxidative stress and inflammation were inseparably linked as they produced a vicious cycle in which oxidative stress triggered inflammation by many mechanisms including activation of the transcription factor kappa B which resulted in the activation and recruitment of immune cells 7 (Figure 1). Inflammation, in turn, triggered oxidative stress through production of reactive oxygen species and reactive nitrogen species by the activated leukocytes and resident cells. Together these events promote tissue damage by inflicting apoptosis, necrosis and fibrosis 85.
Under physiological conditions, oxidative stress gives rise to upregulation of the endogenous antioxidant and cytoprotective proteins and enzymes to prevent tissue injury. This process was mediated by the activation of the Nrf2 which regulated the basal activity and coordinated induction of numerous genes that encode various antioxidant and phase 2 detoxifying enzymes and related proteins 81. Nrf2 is an inactive complex in the cytoplasm by the repressor molecule, Keap1 which facilitated its ubiquitination (Figure 1). Keap1 contained reactive cysteine residues which function as intracellular redox sensors. Nuclear translocation of Nrf2 occurred by phosphorylation of its threonine or serine residues via upstream kinases, such as mitogen-activated protein kinases, protein kinase C, phosphatidylinositol-3-kinase/Akt, casein kinase-2 and PKR-like ER kinase 78. Regulation of cellular antioxidant and anti-inflammatory machinery by Nrf2 plays a central part in defense against oxidative stress. A number of studies have reported that the imbalance between NF-κB and Nrf2 pathways contributed to CKD and renal fibrosis 63, 77, 80, 81, 86.
Besides, inflammation could in the activation of immune cells, including macrophages, dendritic cells and T cells. These immune cells release profibrotic cytokines and growth factors that contribute to renal fibrosis 87, 88.
Hypoxia and Renal Fibrosis
The kidney was physiologically hypoxic despite its plentiful blood supply, because an oxygen shunt is present between arteries and veins. Therefore, it is reasonable to consider that erythropoietin-producing cells reside in the kidney, where they can sensitively detect hypoxia owing to anemia 89, 90, 91. Physiological hypoxia has been uncovered in mammals and in hypoxia-monitoring transgenic mice and rats produced by using hypoxia-inducible factor system. Expansive kidney hypoxia in CKD has also been verified in both patients and animal models. In CKD, hypoxia appeared in tubulointerstitium via multiple mechanisms. First, glomerulosclerosis resulted in a reduction of flow in downstream peritubular capillary, which further compromised by constriction of efferent arterioles of glomeruli and peritubular capillary owing to RAS activation. Second, the loss of peritubular capillaries occurred owing to fibrosis reduced blood perfusion. Third, excessive ECM deposition by fibrogenesis increased the distance between capillary and tubular, diminishing the efficiency of oxygen diffusion.
Upregulation of hypoxia-inducible factor 1α (HIF-1α) in transgenic mice upregulated vascular endothelial growth factor and platelet-derived growth factor-B expression and augmented endothelial cell proliferation. Although increased production and decreased deposition of ECM were observed in transgenic mice compared to control mice, they did not display renal injury or dysfunction 92. These results were consistent with study indicating that conditional knockout of HIF-1α in the proximal tubules lessened fibrosis in mouse UUO 89. Given that deposition of ECM was a part of repair processes unless it is uncontrolled, hypoxia-inducible factor activation by hypoxia in tubular cells mitigated renal injury by the upregulation of angiogenic and fibrogenic factors.
Uremic Solutes and Renal Fibrosis
Fibrosis was the final result of a complex signaling cascade of intracellular and intercellular and molecular responses initiated by organ injury 10. The fibrotic process and fibrotic-associated pathways are conserved between different organs. EMT has emerged as a mainly origin of collagenous matrix-producing myofibroblasts that contributed to the fibrotic response 46, 93, 94, 95. Renal fibrosis ends in uremic stage, yet uremia per se also further promoted the fibrogenesis owing to the direct biological effects of uremic toxins, such as, indoxyl sulfate (IS) and p-cresyl sulfate (p-CS). At least five uremic toxins showed a direct link to EMT and renal fibrosis 96, 97, 98, 99.
Uremic toxin IS was a small organic aromatic polycyclic anion derived from dietary tryptophan by gut microbiota that has widely been investigated in linking with CKD-associated cardiovascular disease 96, 100, 101, 102, and IS can induce vascular calcification and correlates with coronary artery disease and mortality 103. IS also contributed to a plethora of pathologies observed in dialysis patients, including tubulointerstitial inflammation and kidney damage 96. IS overload augmented the gene expression of tissue inhibitor of metalloproteinases-1, intercellular adhesion molecule-1, alpha-1 type I collagen, and TGF-β in the renal cortex of 5/6 nephrectomized rats 104. Moreover, IS stimulated the production of TGF-β in renal proximal tubular cells. Other study indicated that stimulation of HK-2 cells to IS resulted in a reactive oxygen species-mediated upregulation of plasminogen activator inhibitor-1, a downstream signaling mediator of the TGF-β signaling related to most aggressive kidney diseases 105. Furthermore, another study demonstrated that IS can increase α-SMA and TGF-β expression in HK-2 cells by activation of the (pro)renin receptor through reactive oxygen species-Stat3-NF-κB pathways 106. IS also activated the TGF-β signaling, as showed by an increased Smad2/3 phosphorylation 97, 107.
Although EMT contribution to fibrosis was controversial, phenotypic alterations reminiscent of EMT, also presented as epithelial phenotypic changes, might play an important role in the fibrogenesis and disease progression 108. A number of studies have demonstrated that IS induced EMT, as indicated by a downregulated expression of E-cadherin and zona occludens-1, and upregulated α-smooth muscle actin (α-SMA) expression in rat proximal tubular cells (NRK-52E) and rat kidneys 109. Furthermore, IS promoted EMT-associated transcription factor Snail expression, concurrent with an elevated expression of α-SMA and fibronectin and diminished E-cadherin expression in vitro97. Similar effects of IS have also been observed in human renal cell models 109.
In addition, genetic or microRNA-based mechanisms are also reported to inhibit renal fibrosis through modulating signaling pathways to prevent the progression of renal fibrosis during CKD. Knockdown of profibrotic factor Smad4 alleviated renal fibrosis in mice 110. microRNA-23b, microRNA-30e and microRNA -135a was significantly altered in CKD mice 111, 112, indicating microRNAs as biomarkers and therapeutic targets for CKD.
Conclusion
From the above it is clear that our knowledge of action mechanisms contributing to renal fibrosis had rapidly investigated and expanded over the several decades but we were still confused. Nevertheless, the novel knowledge obtained recently points to many new methods to combat renal fibrosis, at least partial reversal of fibrotic tubulointerstitial injury. Future investigation need to clarify whether individual mechanism contributes to all or at least many renal fibrosis models and would therefore be main candidates for therapeutic strategy and intervention at very early stage of fibrogenesis.