
Stereoselective Synthesis of N-Glycosyl Oxazolines and Evaluation of Their Antiproliferative Activity
Abstract
A stereoselective synthesis of protected N-glycosyl oxazolines has been developed from available acylated sugar 1,2-O-acetonides using intramolecular Ritter-like reactions. New N-α- and β-D-pentofuranosyl, α-D-hexofuranosyl oxazolines as valuable intermediates for preparation of diverse N-glycosides were obtained by BF3.OEt2-KHF2 or BF3.OEt2-promoted reactions of pentofuranose and hexafuranose acetonide derivatives with nitriles. When selectively acylated D-xylo- or ribofuranoses were employed in the reactions, N-α-pentofuranosyl oxazolines were prepared in good yields. A mechanism for the formation of glycosyl oxazolines was proposed. A series of oxazoline derivatives were evaluated for their antiproliferative activity on three human cancer cell lines (MCF-7, Hela and K562).
Article Information
- Received
- Accepted
- Published
Academic Editor: Karunamoorthy Jayamoorthy, St. Joseph's College of Engineering.
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2023 Grigorii Sivets, et al.
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Corresponding author: Grigorii Sivets, Institute of Bioorganic Chemistry, National Academy of Sciences of Belarus, 220084 Minsk, Acad. Kuprevicha 5/2, Belarus —
Competing Interests
The authors have declared that no competing interests exist.
Funding
No specific funding statement was provided by the authors.
Data Availability
No data-availability statement was provided by the authors.
Acknowledgements
This study was supported by grants from Belarusian Fond Fundamental Investigations (X-16-048) and FOI «Chemical processes, reagents and technologies, bioregulators and bioorgchemistry», s/p «Chemical foundations of life activity processes» (Bioorgchemisrty 2.3.2.2).
Citation:
Introduction
2-Oxazolines belong to an interesting class of heterocyclic compounds with versatile synthetic applications 1, 2. Carbohydrate-fused oxazolines with a C1-O-linkage have found significant use for the chemical and enzymatic synthesis of oligosaccharides 3and glycoconjugates 4, the preparation of modified carbohydrates, and the design of synthetic oligoamidosaccharides through cationic ring-opening polymerization 5. It is worth noting that isomeric C1-N linked N-glycosyl oxazolines are of special interest in carbohydrate chemistry and these molecules have been used as valuable intermediates in constructing different N-glycoproteins6. However, only a few synthetic routes have been reported to produce isomeric N-glycosyl oxazolines (Scheme 1). Garcia Fernandez and co-workers explored conversions of β-d-fructopyranose and d-fructofuranose 1,2-O-acetonide derivatives (I) with various nitriles in the presence of triflic acid to obtain spiro glycosyl oxazolines (II) 7 by Ritter-like transformations (Scheme 1). Vangala and Shinde synthesized spiro 2-substituted 2-oxazolines ribosides (II) in good yields from 1,2;3,4-di-O-acetonide β-d-psicofuranose derivatives, using stereoselective TMSOTf-mediated Ritter-like reactions with nitriles 8. One-pot syntheses of different protected N-glycooxazolines (IV a,b) and - glycoaminooxazolines (IV c) of interest as potential inhibitors of glycosidases and chitinases have been developed by De Castra et al. via reactions of benzyl- and TBDMS-protected d-glucals with various amides in the presence of N-iodosuccinimide (Scheme 1b) 9. In addition, syntheses of the protected glucopyranosyl oxazoline (IVa) were also investigated from glucopyranosyl azides (e.g., V) 6, 10, 1,2-anhydroglucopyranose derivative (VI) prepared by oxidation of glucal (III) 11, but there is still need for development of practical and efficient routes to various furanosyl or pyranosyl oxazolines containing the C1-nitrogen linkage.
Scheme 1. Stereoselective synthetic routes to N-glycosyl oxazolines from different carbohydrate precursors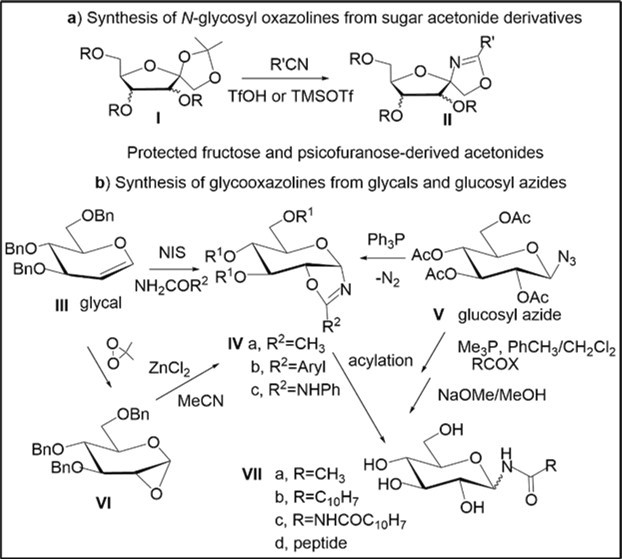
Download figure
Carbohydrate-based N-glycosyl oxazolines have been employed as precursors in stereoselective syntheses of glycosyl isothiocyanates and amides. Acylated α-glycopyranosyl isothiocyanate was synthesized from N-glycooxazoline using copper (II) chloride as additive, and the similar ring-opening reaction of the glycooxazoline precursor with thiophosgene afforded β-glycopyranosyl isothiocyanate in the absence of any additive 12. N-α-Glycosyl amides and N-α- or β-glycopeptides were obtained from azide V throughthe formation of the intermediate glucopyranosyl oxazoline (IVa) followed by α- or β-acylation 6, 13. The stereoselective approach to diverse N-β-glycosyl amides (VII) was developed via a PMe3 mediated Staudinger reaction of glycopyranosyl azides (e.g. V) with carboxylic acid derivatives 13, 14 (Scheme 1). This paper reports a convenient and efficient method towards various N-furanosyl oxazolines based upon BF3.OEt2-promoted Ritter-like reactions of protected sugar derivatives and evaluation of the antiproliferative activity of acylated N-glycosyl oxazolines.
Results and Discussion
Synthesis of N-glycosyl oxazoliones from sugar 1,2-O-acetonides and selectively protected D-pentofuranose deivatives
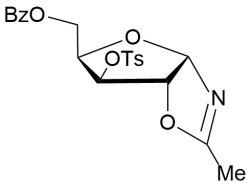
During investigation of different approaches towards fluorodeoxy d-pentofuranoses we have found that reaction of the 3-O-p-toluenesulfonyl xylofuranose derivative 3, prepared via diacetonide 115 from d-xylose, witha3.5-foldaccess of the complex of KHF2with dibenzo-18-crown-6 in acetonitrile in the presence of BF3.Et2O resulted in the formation of N-xylofuranosyl oxazoline7 after the basic aqueous work-up of the reaction mixture and chromatography on silica gel. A selective transformation of the xylofuaranose acetonide derivative 3 with the solvent was observed at the 1,2-O-isopropylidene group in the presence of the Lewis acid (6-7.0 equiv) without formation of fluorinated products by a nucleophilic substitution reaction of the 3-O-p-toluenesulfonyloxy group with inorganic fluoride (Scheme 2).However, application of crown ether gave rise to tedious purification of the product by column chromatography. No reaction was observed under treatment of the tosylate 3 with a 3.5-foldaccess of KHF2 in CH3CN at rt and only the startingacetonide was recovered unchanged. Further, it was shown that the oxazoline 7 can be prepared from the acetonide derivative 3 in a high 98% yield using BF3.Et2O/KHF2 in CH3CN without column chromatography on silica gel (Scheme 2) as compared to the previous findings reported earlier 16.
Scheme 2. Synthetic study of N-glycosyl oxazolines from sugar acetonides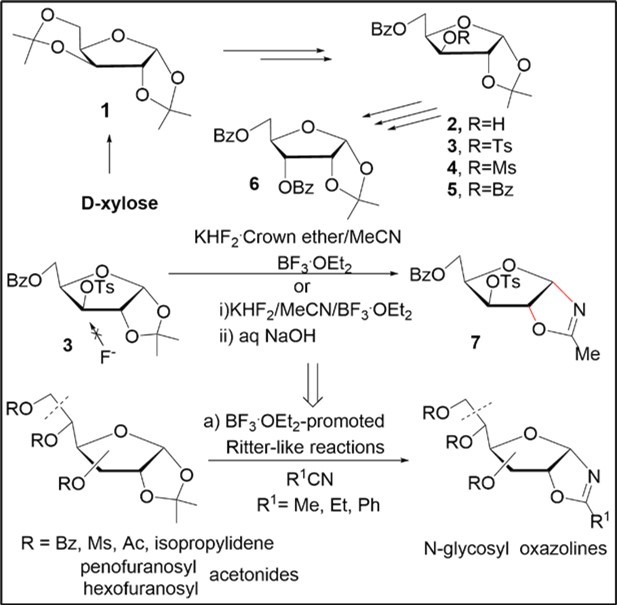
Download figure
In the course of present comprehensive study, conversions of various protected d-pentofuranose and -hexofuranose acetonides with nitriles to glycosyl oxazoline derivatives were explored under BF3.Et2O/KHF2 reaction conditions at room temperature (Scheme 2 and Table 1). The reactionof the 3-O-mesyl xylofuranose derivative 4 gave the oxazoline 8 in 93% yield without formation of nucleophilic substitution products as with the tosylate 3. N-Pentofuranosyl oxazolines 9, 13 and 16 were synthesized in high 96-99% yields in acetonitrile (Table 1, entries 3,7 and 9) from isomeric benzoylated 1,2-O-isopropylidene-d-pentofuranose derivatives 5-6, and 15,prepared by the known methods decribed earlier from d-xylose and arabinose 17, 18, 19, 20, 21. 1,2;3,5-Di-O-isopropylidene-d-xylofuranose (1) also afforded the protected xylofuranosyl oxazoline 12 in 76% yield as the result of regioselective transformations in the 1,2-O-isopropylidene group (Table 1, entry 6). The reactions of benzoyl-protected d-xylofuranose, ribofuranose and arabinofuranose 1,2-O-acetonides studied in acetonitrile at room temperature gave rise to the stereoselective formation of cis-fused bicyclic N-α- and β- d-pentofuranosyl oxazolines after the work-up of the reaction mixture without using column chromatography on silica gel (Table 1, entries 1-3, 6,7 and 9). Further, we explored scope of the BF3.Et2O-KHF2-mediatedreaction of benzoylated 1,2-O-isopropylidene-d-pentofuranose derivatives with other nitriles such as propionitrile and benzonitrile. The reaction 6 with benzonitrile or propionitrile gave oxazolines 10 and 11 in 97% and 86% yields, respectively (entries 4 and 5). The protected α-ribofuranosyl and β-arabinofuranosyl oxazolines 14 and 17 were smoothly prepared from the Ritter-like reactions of acylated 1,2-O-acetonides of 6 and 15 in benzonitrile in 97% yield (Table 1, entries 8 and 10). Next, the above stereoselective reactions were investigated for acyl-protected hexofuranose 1,2-O-acetonide derivatives under the similar conditions. 3,5,6-Tri-O-benzoyl-1,2-O-isopropylidene-α-d-glucofuranose (18) as well as isomeric allofuranose derivative 24, prepared according to the known methods 22, gave protected N-glycofuranosyl oxazolines 19 and 20 in acetonitrile and benzonitrile, oxazoline 25 in benzonitrile,respectively, in the high yields (Table 1, entries 11, 12 and 15). The Ritter reaction of fully O-acetylated 1,2-O-acetonide-α-d-glucofuranose 2123 or α-d-allofuranose 2624 in acetonitrile or benzonitrile furnished oxazolines 22 (entry 13), 23 (entry 14) and 27 (entry 16) in 95-98% yields. The structures of synthesized oxazolines were supported by 1H, 13C NMR, IR spectral data and mass spectra (Experimental part).Resonance signals of CH3 groups of oxazoline rings for all synthesized glycosyl oxazolines were observed as singlets in the range of ~ 1.97-2.18 ppm and 13.2-14.2 ppm 7 in 1H and 13С NMR spectra, respectively, measured in CDCl3. Signals of the tertiary carbon atoms of the sugar oxazolines with 2-Me, Et or Ph substituents displayed at 167-173 ppm in 13С NMR spectra. Absorption bands of
Table 1. Synthesis of N-pentofuranosyl and N-hexofuranosyl oxazolines from protected d-sugar acetonides using BF3 OEt2-KHF2-promoted reactions with nitriles| Entry | Protected acetonide | Nitrile | Time (h) | KHF2/BF3.Et2O(Mol equiv) | Product | Yielda (%) |
| 1 |
 3
3
|
CH3CN | 18 | 3.0/7.2 |
 7
7
|
98% |
| 2 |
 4
4
|
CH3CN | 18 | 3.1/7.2 |
 8
8
|
93% |
| 3 |
 5
5
|
CH3CN | 18 | 3.5/6.3 |
 9
9
|
96% |
| 4 |
5
|
C6H5CN | 18 | 3.5/6.2 |
 10
10
|
97%b |
| 5 |
5
|
C2H5CN | 18 | 2.7/4.9 |
 11
11
|
86%c |
| 6 |  1 1 |
CH3CN | 4 | 2.1/6.2 |
 12
12
|
76% |
| 7 |
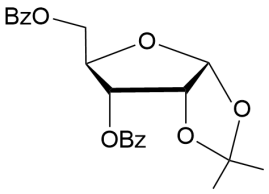 6
6
|
CH3CN | 18 | 3.5/6.3 |
 13
13
|
99% |
| 8 |
6
|
C6H5CN | 18 | 2.7/6.2 |
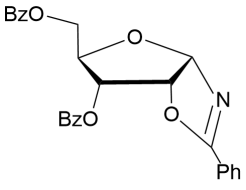 14
14
|
97%b |
| 9 |
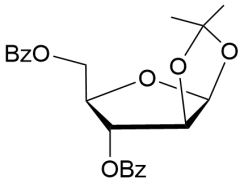 15
15
|
CH3CN | 18 | 3.5/6.3 |
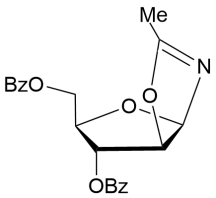 16
16
|
99% |
| 10 |
15
|
C6H5CN | 18 | 4.8/6.3 |
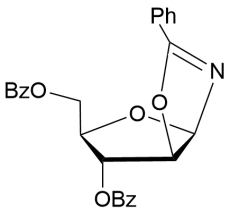 17
17
|
97%b |
| 11 |
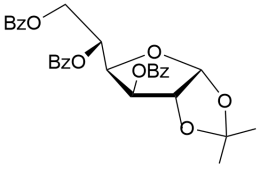 18
18
|
CH3CN | 18 | 3.4/8.4 |
 19
19
|
93% |
| 12 |
18
|
C6H5CN | 18 | 4.9/8.3 |
 20
20
|
92%b |
| 13 |
 21
21
|
CH3CN | 18 | 2.5/8.1 |
 22
22
|
95% |
| 14 |
21
|
C6H5CN | 18 | 4.9/8.3 |
 23
23
|
92%b |
| 15 |
 24
24
|
C6H5CN | 18 | 4.3/8.3 |
 25
25
|
86%b |
| 16 |
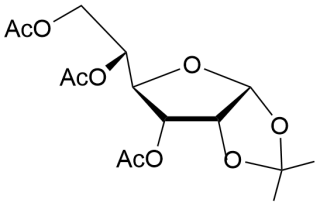 26
26
|
CH3CN | 18 | 2.4/8.0 |
 27
27
|
98% |
Thus, we have found that the Ritter-like reactions 25, 26 of the protected pentofuranose and hexofuranose 1,2-O-acetonides with nitriles in the presence of BF3.Et2O and KHF2 resulted in stereoselective transformations in the 1,3-dioxolane ring to give the only reaction products containing the five-membered 2-oxazoline derivatives. Next, to understand the assumed role of KHF2 as a promoter with acidic properties in the studied conversions of acetonides 1, 3 and 5 into glycosyl α-D-oxazolines in the presence of BF3.Et2O,the Ritter-like reaction of benzoylated d-xylofuranose 1,2-O-acetonide 5 was testedwith acetonitrile in the presence of 7.2 equiv of BF3.Et2O and 3.4 equiv of p-toluenesulfonic acid instead of KHF2 at room temperature (Scheme 3, conditions a).
The oxazoline 9 was prepared in 78% yield after column chromatography on silica gel.Besides, the BF3.Et2O-KHF2-assisted reaction of the tosylate 3 in acetonitrile was studied in the presence of NaBH4 at room temperature (Scheme 3, conditions b). Such treatment of compound 3 did not result in the reduction of the C3-O-p-toluenesulfonyloxy group and acyclic product 30 was obtained in 40% yield after chromatography on silica gel. The structure of the xylitol derivative 30 was confirmed by the preparation of fully O-benzoylated derivative 31 (70%) and analysis of their 1H and 13C NMR, HRMS spectral data. Proposed synthetic pathway to compound 30 via the possible reductive cleavage of the ketal-protected xylofuranose derivative 3 is outlined in Scheme 3. Activation of the 1,3-dioxolane ring in 3 may proceecd in the presence of the Lewis acid BF3 or an acidic promoter with generation of intermediate 29 in the first step. Then, the formation of an intermediate oxocarbenium ion 29 occurs from 28. A selective reduction of aldofuranose counterpart of 29 with diborane forming in situ from NaBH4 and BF3 yields the selectively protected xylitol derivative 30 in the next steps (Scheme 3).The above findings indicate that the BF3.Et2O-mediated reactions of benzoylated d-pentofuranose 1,2-acetonides imply a regioselective activation of the 1,2-O-isopropylidene group with involvement of the Lewis acid and acid promoters such as KF.HF or p-TsOH, as with the Ritter-like reactions described for the fructofuranose acetonides in the presence of triflic acid 7or natural monosaccharides in liquid HF 27.
Scheme 3. Study of BF3.OEt2-assisted transformations of protected xylofuranose acetonides 3 and 5 on the 1,3-dioxolane ring in acetonitrile. Reagents and conditions: (a) 5, CH3CN, p-TsOH, BF3.Et2O, rt, 18 h; 1N aq NaOH, 9, 78%; (b) 3, KHF2/BF3.Et2O, NaBH4, CH3CN, rt, 5% aq NaHCO3, 40%, 30; (c) BzCl, Py, rt, 31, 65%.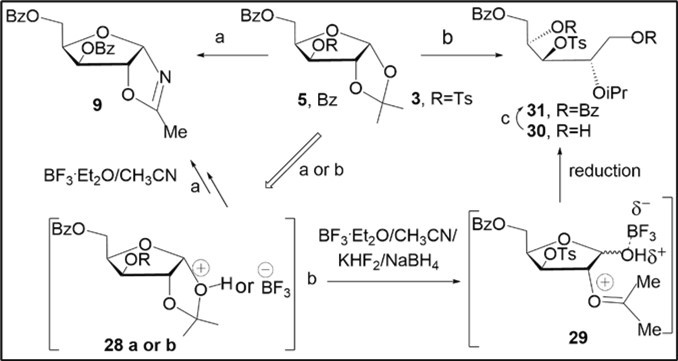
Download figure
To further explore Ritter reactions, syntheses of the protected N-glycosyl oxazolines were investigated from the d-pentofuranose 1, 3, 5 and hexofuranose 18, 21, 24 acetonide derivatives under various Lewis acid-assisted conditions (Table 2).
The control Ritter reaction of xylofuranose acetonides 3 or 5 was tested in acetonitrile in the presence of catalytic amounts of BF3.Et2O (0.5-0.8 equiv) and the excess of KHF2 (3.5 equiv). No formation of oxazolines 7 and 9 was observed under these conditions (Table 2, entries 1 and 2). The reactions of acetonides 3 and 5 did not proceed in MeCN containing the excess of KHF2 ora complex of KHF2 with 18 crown 6 prepared previously in anhydrous methanol (entries 3 and 4). It was found that treatment of acetonides 3 and 5, unlike diacetonide 1 (entry 9), with 7.2 and 6.3 equiv of BF3.Et2O in dry acetonitrile without KHF2 led to oxazolines 7 and 9 in high 93% and 92% yields, respectively (entries 7 and 8).
Table 2. Screening in Ritter-like reactions of protected xylofuranosyl, glucofuranosyl and allofuranosyl acetonides with nitriles under the Lewis acid activated conditions| Entry | Protectedacetonide | Reaction conditions | Oxazoline(yield,%)a |
| 1 | 3 | BF3.Et2O (0.5 equiv)/CH3CN/KHF2 (3.5 equiv), rt, 18 h | - |
| 2 | 5 | BF3.Et2O (0.8 equiv)/CH3CN/KHF2 (3.5 equiv), rt, 18 h | - |
| 3 | 3 | CH3CN/ KHF2 (5.6 equiv).18 crown 6, rt, 18 h | - |
| 4 | 5 | CH3CN/KHF2 (3.5 equiv), rt, 18 h | - |
| 5 | 5 | BF3.Et2O (2.0 equiv)/CH3CN/KHF2 (3.5 equiv), rt, 18 h | 9 (20%) |
| 6 | 5 | BF3.Et2O (3.0 equiv)/CH3CN/KHF2 (3.5 equiv), rt, 18 h | 9 (87%) |
| 7 | 3 | BF3.Et2O (7.2 equiv)/CH3CN, rt, 18 h | 7 (93%) |
| 8 | 5 | BF3.Et2O (6.3 equiv)/CH3CN, rt, 18 h | 9 (92%) |
| 9 | 1 | BF3.Et2O (6.2 equiv)/CH3CN, rt, 3 h | 12 (54%)b |
| 10 | 5 | BF3.Et2O (6.3 equiv)/PhCN, rt, 18 h | 10 (91%)b |
| 11 | 5 | BF3.Et2O (4.9 equiv)/EtCN, 00→ rt, 18 h | 11 (72%) |
| 12 | 18 | BF3.Et2O (7.5 equiv)/PhCN, rt, 18 h | 20 (44%)b |
| 13 | 21 | BF3.Et2O (7.6 equiv)/PhCN, rt, 18 h | 23 (40%)b |
| 14 | 24 | BF3.Et2O (7.8 equiv)/PhCN, rt, 18 h | 25 (50%)b |
| 15 | 3 | TMSOTf (7.2 equiv)/CH3CN/KHF2 (3.5 equiv), rt, 18 h | 7 (98%) |
| 16 | 3 | TMSOTf (7.2 equiv)/CH3CN, rt, 18 h | 7 (95%) |
Ritter reactions of benzoylated xylofuranosyl acetonide 5 with benzonitrile or propionitrile in the presence of6.0 or 4.9 of equiv BF3.Et2O resulted in oxazolines 10 and 11 in 91% and 72% yields (entries 8 and 9). The Ritter reaction of O-benzoylated or acetylated 1,2-O-acetonide-d-glucofuranose derivatives 18 and 21 with benzonitrile in the presence of BF3.Et2O (7.5 equiv) gave the oxazolines 20 and 23 in 44% and 40% yields (entries 12 and 13) compared to the same reactions under the BF3.Et2O-KHF2-promoted conditions (92% and 95%) (Table 1, entries 12 and 13). The allofuranosyl oxazoline derivative 25 was prepared in high yields using the BF3.Et2O-KHF2- or BF3.Et2O conditions forRitter reactions of the D-allofuranose acetonide 24 with benzonitrile in the presence of 7.5 equiv of the Lewis acid (Table 2, entry 14 and Table 1, entry 15). Ritter reactions of the acetonide 3 with acetonitrile have been tested using the TMSOTf-KHF2 or TMSOTf-mediated conditions (entries 15 and 16) and the oxazoline 7 was prepared in 98% and 95% yields, respectively.
After screening of various BF3.Et2O-promoted reactions of xylofuranose acetonides 1, 3, and 5 with different protecting groups we have found that benzoyl-protected N-glycosyl oxazolines can be prepared in good yields under the BF3.Et2O-KHF2 (Table 1) or BF3.Et2O (Table 2) conditions in the presence of the excess of the Lewis acid. In the case of a series of acylated hexofuranose 1,2-O-acetonide derivatives 18, 21, 24 and 26, the excess of the Lewis acid (about 8 equiv) along with KHF2 (2.5-4.0 equiv), that may generate HF or HBF4 and KBF4 after interaction with the strong Lewis acid in polar solvent, needs for conversions of acetonides into oxazolines under the BF3.Et2O-KHF2 (Table 1, entries 11-16) with good yields compared to the BF3.Et2O-promoted reactions for glucofuranose acetonides 18, 21 andallofuranose acetonide 24 (Table 2, entries 12-14). It is important to note that selection of optimal conditions (the use of the acidic promoter, ratio of reagents, excess of LW) for achieving high yields of glycosyl oxazolines in the Ritter reactions under consideration depends on the structure of the starting sugar, a character of protecting groups and nitrile used as solvent/reagent. Based on analysis of different conditions explored for a series of the Ritter-like reactions of sugar acetonides, mechanistic pathways leading to the formation of N-α-glycosyl oxazolines from protected xylofuranose acetonides 1, 3 and 5 were proposed (Scheme 4. and Scheme 5). Proposed mechanism for stereoselective BF3.Et2O-KHF2-assisted reactions of xylofuranose acetonide derivatives 3 and 5 with nitriles is illustrated in Scheme 4.
Scheme 4. Proposed mechanism for the formation of oxazolines from d-xylofuranose acetonide derivatives 3 and 5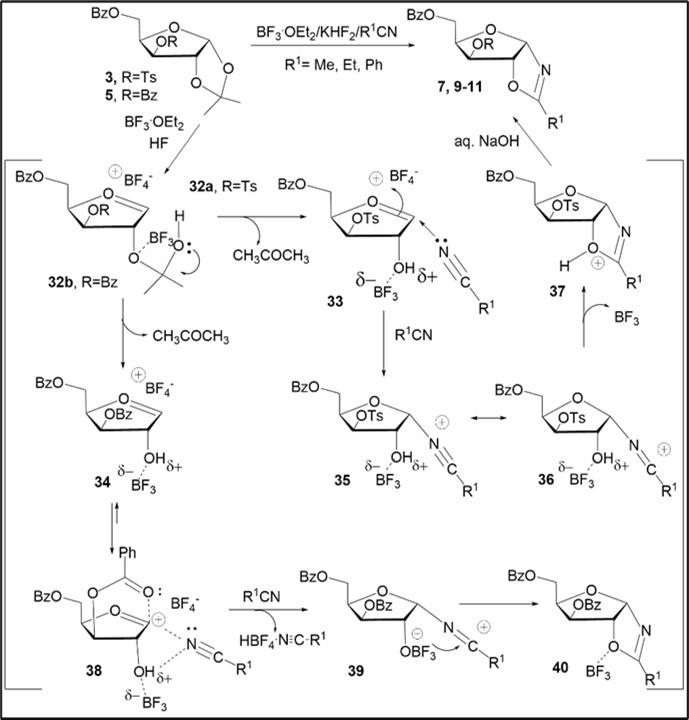
Download figure
The synthetic route to protected N-α-xylofuranosyl oxazolines likely to include the formation of intermediate ions 32a and 32b with assistance of a mild acidic promoter (gradual generation from KHF2 in the presence of excess of BF3.Et2O in polar solvent) and the Lewis acid, and the subsequent occurrence of oxocarbenium ions 33 and 34, respectively 12, 27. We suggest that mechanistic pathway towards the nitrilium intermediate 35 from tosylate3may occur through a direct nitrile addition to the furanosyl oxocarbenium ion 33 from α or β-face and without remote participation of the protecting groups. The formation of the thermodynamically more stable the α-nitrilium ion 35 as compared with an intermediate β-nitrilium ion is probably favored by activated with the Lewis acid the 2-hydroxyl group, which is capable of stabilizing the adjacent cation via interaction with the α-nitrilium group in the presence of BF3. Notice that the preferential generation of an intermediate stable α-nitrilium ion under the conversion of the protected xylofuranose derivative in CD3CN in the presence of the Lewis acid (Me3OBF4) has been supported by Turnbull and co-workers using 1H NMR experimental data and DFT calculations 28.The kinetically controlled formation of the α-xylofuranosyl nitrilium ion 35 can result from the oxocarbenium ion 33 or contact tetrafluoroborate ion pairs via solvation of the intermediate oxonium cation under SN1-reaction and a fast attack with nitrile from the α-face due to an anomeric effect, as has been reported for the glycosylation reactions of pyranose derivatives through pyranosyl nitrilium ions 29, 30, 31, 32. The generation of oxazolinium intermediate 3729 proceeds from the cation 36 via intramolecular trapping of the 2-O-hydroxyl group with the electrophilic nitrilium carbon.
Another possible pathway for formation of acylated N-α-xylofuranosyl oxazolines via generation of intermediate cyclic benzoxonium ions with participation of acyl protecting groups should be considered in the case of BF3.Et2O-KHF2-assisted reactions of benzoylated D-xylofuranose 1,2-O-acetonide5. α-Xylofuranosyl nitrilium intermediates may arise by nitrile addition to the intermediate cyclic 1,3(1,5)-dioxacarbenium ions, which would be produced via assistance of the O-benzoyl groups in the oxocarbenium ion 34 in the presence of the Lewis acid. The influence of vicinal and remote O-acyl groups has been invoked on many glycosylation reactions of monosaccharide derivatives in the presence of Lewis acids32, 33, 34. The remote stereodirecting participation of 3-O- or 4-O-acyl (benzoyl, 4-methylbenzoyl or acetyl) protecting groups and their distinct stereochemical effects for promoted glycosylation reactions of protected pyranoses and furanosesas glycosyl donors have earlier been examined 34, 35. An interesting concept of catalysis for the glycosylation reactions was reported which was introduced by the Schmidt group 36, 37, 38. It includes activation of acceptor and glycosyl donor in the presence of Lewis acids as catalysts followed by generation of a cyclic intermediate to give rise to O-glycoside(s) as a result of the stereoselective glycosidation. From those mechanistic considerations, pathway for the BF3.Et2O-KHF2-promoted reaction ofacetonide5wasproposed (Scheme 4). One may pass through an intermediate transition state or complex 38 that originates from coordination of a transient glycosyl cation, stabilized by the remote participation of 3-O-benzoyl group in the oxocarbenium ion, with acetonitrile under assistance of the 2-OH group activated in the presence of BF3.Et2O as a strong Lewis acid 32, 37.
The further stereoselective course of the Ritter reaction ofacetonide 5would result inα-xylofuranosyl nitrilium ions39 and generation of an adduct 40 as a complex of the oxazoline with BF3.The basic work-up of intermediate oxazolinium derivatives 37 and 40 with aqueous sodium hydroxide gave protected N-α-xylofuranosyl oxazolines 7, 9-11 (Scheme 4).
Two different pathways for BF3.Et2O-promoted Ritter-like reactions of d-xylofuranose diacetonide 1, bearing non-participating isopropylidene groups, are shown in Scheme 5.
Scheme 5. Proposed intermediates during BF3.Et2O-promoted reactions of diacetonide 1 with acetonitrile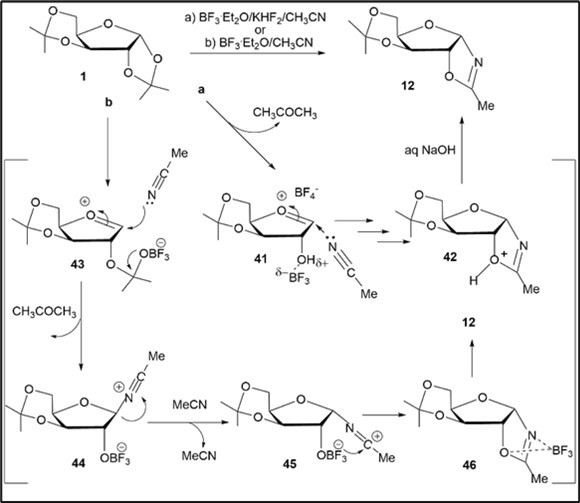
Download figure
The formation of the oxazoline 12 from diacetonide 1 in acetonitrile under the BF3.Et2O-KHF2-assistedconditions (a) may proceed via the oxocarbenium ion 41 after a regioselective activation of the 1,2-O-isopropylidene group with an acidic promoterand the Lewis acid followed by generation of the oxazolinium intermediate 42 similar to conversions of the 3-O-tosyl xylofuranose derivative 3 (Scheme 4) into the oxazoline 7 throughthe cation 36 and key oxazolinium derivative 37. Under the BF3.Et2O-mediatedconditions (b), the Ritter-like reaction of 1 with acetonitrile would occur in a different pathway through activation of the 1,3-dioxolane ring with BF3.Et2O in the first step, generation of the oxocarbenium ion 43 and a subsequent bottom attack of solvent to give the β-nitrilium intermediate 44, as has been reported for the preparation of oxazolines by reacting epoxides with nitriles in the presence of Lewis acids 11, 39. The further inversion of 44 at C1 with acetonitrile would result in an intermediate α-nitrilium ion, and subsequently the electrophilic α-nitrilium cation 45, a complex of the oxazoline with the Lewis acid 46, giving the target oxazoline 16 after the basic work-up.
In order to explore the scope of the BF3.Et2O-mediated approachfor other protected d-pentofuranose derivatives with free hydroxyl groups we have undertaken synthesis of N-xylofuranosyl oxazolines from selectively benzoylated xylofuranoses 47-48 readily prepared by the acidic removal of 1,2-O-isopropylidene groups from xylofuranose acetonide derivatives 2 and 5 with aqueous trifluoroacetic acid (Scheme 6, conditions a1-2). 5-O-Benzoyl-α,β-d-xylofuranose (47) gave the benzoyl-protected oxazoline 49 (75%) under the the KHF2-BF3.Et2O conditions (conditions b1). Interestingly, the reaction of 3,5-di-O-benzoyl-α,β-d-xylofuranose (48) in acetonitrile furnished the oxazoline 9 (99%), as in the case of the Ritter reaction of 3,5-di-O-benzoyl-1,2-O-isopropylidene-α-d-xylofuranose (5) (Table 1, entry 3).
In addition, the Ritter-like reaction of 48 with acetonitrile in the presence of BF3.Et2O (6.0 equiv) without KHF2 (conditions c1) also afforded the protected N-α-xylofuranosyl oxazoline 9 (65%). Furthermore, we have found that the Lewis acid promoted reactions of 1,3,5-tri-O-benzoyl-α-d-ribofuranose (50)40 with CH3CN in the presence of KHF2 or without the inorganic salt gave the α-oxazoline 13 in 99% and 65% yields, respectively, after the basic work-up of reaction mixtures (Scheme 6, conditions b2 and c2). Removing benzoyl protecting groups in the oxazoline 17 with NH3/MeOH (conditions d) resulted in the β-arabinofuranosyl oxazoline 51 (93%). Acetylation of the latter with acetic anhydride in pyridine at room temperature afforded fully O-acetylated oxazoline 52 in 80% yield.
Scheme 6. Synthesis of acylated d-xylo-, ribo- and arabinofuranosyl oxazolines. Reagents and conditions: (a1) 2, 93% aq TFA, rt, 2 h, 47, 80%; (a2) 5, 93% aq TFA, rt, 2 h, 48, 80%; (b1) benzoylated d-xylofuranoses 47-48, CH3CN, KHF2,BF3.Et2O, rt; 3-4 h, 5 % aq NaHCO3, 49, 75%; 9, 99%; (c1) 48, CH3CN/ BF3.Et2O, rt, 3 h, 5 % aq NaHCO3,9, 65%; (b2) 50, CH3CN, KHF2, BF3.Et2O, rt; 3 h, 1N aq NaOH,13, 99%; (c2) 50, CH3CN, BF3.Et2O, rt, 3 h, 1N aq NaOH,13, 65%; d) 17, NH3/MeOH, rt, 18 h, 51, 93%; e) 51, Ac2O, Py, rt, 52, 80%.
Download figure
From the above-considered synthetic routes to oxazolines from protected d-pentofuranose derivatives it should be noted that the BF3.Et2O-promoted reactions of selectively acylated d-xylofuranose and -ribofuranose derivatives (Scheme 6), diacetonide 1 (Scheme 5) and acylated 1,2-O-isopropylidene-α-d-pentofuranoses 3, 5, 6 and 15 (Scheme 4 and Scheme 3) differ in producing intermediate oxocarbenium ions in the presence of the Lewis acid in the first steps and, probably, different oxazolinium intermediates in the next steps. The formation of α-nitrilium ions is the general peculiarity for the Ritter-like transformations of d-xylofuranose and -ribofuranose derivatives studied with nitriles. Prepared sugar oxazolines can be used for obtaining various N-glycosyl amides via hydrolysis reactions and novel N-glycoside derivatives.
In vitro antiproliferative activity of N-glycosyl oxazoline derivatives with 2-phenyl substituent
A series of newly synthesized N-glycosyl oxazoline derivatives with 2-phenyl substituent were tested for their in vitro inhibitory effects on proliferation of myelogenous leukemia (K562), cervical carcinoma (Hela) and breast carcinoma (MCF-7) using the resazurin assay 41 that, together with other high-throughput screening methods, had been developed previously to measure viability or cytotoxicity 42. 5-Fluorouracil (5-FU) was used as the reference compound. The findings are listed in Table 3.
Table 3. Antiproliferative activities of a series of N-glycosyl oxazoline derivatives with 2-phenyl substituent on human cancer cell lines| Compound | IC50a (ϻM) | ||
| MCF-7 | K-562 | Hela | |
| 10 | 99.76±0.45 | > 100 | > 100 |
| 14 | > 100 | 23.01±0.31 | 63.92±0.21 |
| 17 | 79.4±0.34 | > 100 | > 100 |
| 2 0 | > 100 | > 100 | 21.92±0.25 |
| 51 | NI | NI | NI |
| 52 | > 100 | > 100 | > 100 |
| 5-FU | 26.32±0.32 | 11.02±0.26 | 32.43±0.17 |
Among N-pentofuranosyl oxazolines with xylo-, ribo- and arabino-configurations tested(compounds 10, 14, 17, 51 and 52), only benzoylated xylo- and arabinofuranosyl oxazolines 10 and 17 displayed weak inhibitory effects against MCF cell line with IC50 values of 99.76 and 79.4 ϻM, respectively. Unlike to isomeric xylo- and arabino-furanosyl oxazolines 10 and 17, 3,5-di-O-benzoyl α-ribofuranosyl oxazoline 14 showed moderate activity with IC50 value of 63.92 ϻM in Hela cells and good antiproliferative activity against K562 cell line (IC50 23.01 ϻM). Deprotected N-β-d-arabinofuranosyl oxazoline 51 did not show activity on all cell lines at the highest concentration of tested compound. The benzoylated N-α-glycofuranosyl oxazoline derivative 20,bearing 2-phenyl substituent in the oxazoline ring,displayed significant antiproliferative activity with IC50 value (21.92 ϻM) comparable to those of the knownnucleobase analog 5-FU on Hela cells. Comparative biological assessment of the oxazoline 20 and itsclose structural analogs the N-glycosyl oxazoline derivatives 23 and 25 is underway in cancer cell lines. To gain insight into the mode action/mechanism for the inhibitory effects of the oxazolines with 2-phenyl substituent, the apoptosis assays for compound 20 as well as its two analogs are currently under investigation in Hela cancer cell line, applying DAPI and Annexin V-FITC/PI staining methods, and the results will be published elsewhere.
Conclusion
In summary, a convenient and stereoselective approach to prepare various N-glycosyl oxazolines has been developed from sugar 1,2-O-acetonides using mild reaction conditions for the BF3.OEt2-mediated Ritter-like reactions. Scope of a novel method based upon the reactions of selectively protected d-pentofuranose derivatives with nitriles as solvents in the presence of the excess of the Lewis acid and potassium hydrogen difluoride, or BF3.OEt2-assisted conditions, was examined for the preparation of blocked carbohydrate-based oxazolines. A series of new oxazolines as valuable intermediates to prepare N-glycosyl amides, modified sugars and N-glycopeptides were synthesized in high yields, and screened for their inhibitory effects on proliferation of three human cancer cell lines. Of various 2-phenyl substituted N-furanosyl oxazolines evaluated, only the benzoyl-protected glucofuranosyl and ribofuranosyl oxazoline derivatives were found to exhibit good growth inhibition activities against two different cancer cell lines.
Experimental section
General information. Column chromatography was performed on silica gel 60 H (70-230 mesh; Merck, Darmstadt, Germany), and thin-layer chromatography (TLC) on Merck silica gel aluminum 60 F254 precoated plates. All commercially available reagents were used without further purification. 1H and 13C NMR spectra were recorded in CDCl3 and CD3OD with a Bruker Avance-500-DRX spectrometer at 500.13 and 126.76MHz, respectively. 1H and13C NMR chemical shifts (δ, ppm) are relative to internal chloroform peak (7.26 ppm for 1H and 77.0 for 13C NMR). Splitting patterns were reported as following: s: singlet, d: doublet, t: triplet, m: multiplet. J values are reported in Hz. Optical rotations were measured with Autopol III automatic polarimeter. IR spectra were measured with on PerkinElmer Spectrum 100FT-IR spectrometer. Melting points were determined on a Boetius apparatus and were uncorrected. High resolution mass spectra (HRMS) were recorded on an Agilent Q-TOF 6550 Instrument (USA) using ESI (electrospray ionization).
Synthesis of N-glycosyl oxazolines from protected pentofuranose 1,2-O-acetonides
a1. Synthesis 2-alkyl-α-D-pentofurano-(1,2-d)-2-oxazoline derivatives under the BF3.Et2O-KHF2-promoted conditions. To a stirred solution of pentofuranose acetonide derivative (1.4 mmol) in anhydrous acetonitrile (8.6 ml) or benzonitrile (3.8 ml) KHF2 (4.8 mmol) and boron trifluoride diethyl etherate (1.42 ml,10.3 mmol) were added successively. The resulting solution was stirred at room temperature for 18 h, and then the reaction mixture was poured into cooled 22.6 ml 1N aq NaOH. The aqueous phase was extracted with CHCl3 (3x100 ml). The combined organic extracts were washed with water, dried over anh. Na2SO4, and evaporated to dryness. Oxazolines 7-8, 11-13 were prepared in 76-99% yields, and oxazolines 10, 14, 17with 2-phenyl substituentwere isolated in 97% yield after column chromatography on silica gel using for elution mixtures of hexane-ethylacetate and ethylacetate-methanol 6:1.
a2. Synthesis 2-methyl-α-D-pentofurano-(1,2-d)-2-oxazoline derivatives under the BF3.Et2O-promoted conditions. To a stirred solution of xylofuranose acetonide derivative (0.2 mmol) in anhydrous acetonitrile (1.5 ml) boron trifluoride diethyl etherate (0.2 ml, 1.44 mmol) was added successively. The resulting solution was stirred at room temperature for 18 h, and then the reaction mixture was poured into cooled 5% aq. NaHCO3. The aqueous phase was extracted with CHCl3 (3x50 ml). The combined organic extracts were washed with water, dried over anh. Na2SO4, and evaporated to dryness. Oxazolines 7, 9 and 12 were prepared in 54-93% yields.
Synthesis of N-glycosyl oxazolines from protected hexofuranose 1,2-O-acetonides
a1. To a stirred solution of acylated glucofuranose or allofuranose acetonide (0.4 mmol) in anhydrous benzonitrile (3.8 ml) or acetonitrile (3.1 ml) KHF2 (1.95 mmol) and boron trifluoride diethyl etherate (0.45 ml, 3.3 mmol) were added successively. The reaction mixture was stirred at room temperature for 18 h, and then poured into cooled 7.3 ml 1N aq NaOH. The aqueous phase was extracted with CHCl3 (3x30 ml). The combined organic extracts were washed with water, dried over anh. Na2SO4, and evaporated. Oxazolines with 2-phenyl substituent were isolated by column chromatography on silica gel using for elution mixtures of hexane-ethylacetate 6:1, 4:1, 2:1, and ethylacetate or ethylacetate-methanol 6:1. Oxazolines 19-20, 22-23 and 25, 27 were prepared in 86-98% yields.
2-Methyl-(5-О-benzoyl-3-О-p-toluenesulfonyl-α-D-xylofurano)-(1,2-d)-2-oxazoline (7).
Yield (98%), a colorless oil(methoda1).(α)D20–44.4 (c 0.5, CHCl3). IR (film, CCl4): ν 1725, 1670, 1615, 1375, 1272 cm-1. 1Н NMR (500 MHz, CDCl3) δ = 7.38-7.85 (m, 9H, СОC6H5 and ОSO2C6H4CH3), 6.09 (d, 1Н, J1,2 = 5.4 Hz, Н-1), 5.02 (d, 1Н, J3,4 = 3.0 Hz, Н-3), 4.86 (d, 1Н,Н-2), 4.33 (dd, 1Н, J5,4 = 6.2, J5,5′ = 11.3 Hz, Н-5), 4.22 (dd, 1Н, J5′,4 = 5.7 Hz, Н-5′), 3.98-4.01 (m, 1Н, Н-4), 2.31 (s, 3H, ОSO2C6H4CH3), 1.96 (s, 3H, NCH3). 13C NMR (126 MHz, CDCl3) δ = 167.4 (CN), 164.9 (C=O, СОC6H5), 145.6, 133.4, 130.3, 129.2, 128.5, 127.6 (СОC6H5 andОSO2C6H4CH3), 100.2 (С-1), 88.4 (C-4), 81.1, 74.2 (C-2, С-3), 60.3 (С-5), 21.0 (ОSO2C6H4CH3), 13.2 (NMe).HRMS (ESI+):m/z calcd for C21H21NO7S (M+H)+: 432.1117, found 432.1120; and C21H21NO7SNa (M+Na)+: 454.0936, found 454.0935.
2-Methyl-(5-О-benzoyl-3-О-methanesulfonyl-α-D-xylofurano)-(1,2-d)-2-oxazoline (8).
Yield (93%), a colorless oil (methoda1).(α)D20–35.1 (c 0.8, CHCl3). IR (film): ν 1722, 1675, 1357, 1275 cm-1. 1Н NMR (500 MHz, CDCl3) δ =7.47-8.09 (m, 5H, СОC6H5), 6.22 (d, 1Н, J1,2=5.6 Hz, Н-1), 5.23 (d, 1Н, J3,4 = 3.1 Hz, Н-3), 5.09 (d, 1Н,Н-2), 4.66 (dd, 1Н, J5,4 = 6.3, J5,5′ = 11.8 Hz, Н-5), 4.62 (dd, 1Н, J5′,4 = 3.4 Hz, Н-5′), 4.13-4.16 (m, 1Н, Н-4), 3.16 (s, 3H, ОSO2CH3), 2.12 (s, 3H, NCH3). 13C NMR (126 MHz, CDCl3) δ = 168.8 (CN), 166.1 (C=O, СОC6H5), 133.5, 129.8, 129.3, 128.5, (СОC6H5), 100.8 (С-1), 84.2 (C-4), 77.3, 75.1 (C-2, С-3), 60.8 (С-5), 38.5 (ОSO2CH3), 13.9 (NMe). HRMS (ESI+): m/z calcd for C15H17NO7S (M+H)+:356.0798, found 356.0791; and C21H21NO7SNa (M+Na)+: 378.0610, found 378.0511.
2-Methyl-(3,5-di-О-benzoyl-α-D-xylofurano)-(1,2-d)-2-oxazoline (9).
Yield (96%), foam (method a1). (α)D20–73.2 (c 1.0, CHCl3). IR (KBr): ν 1722, 1672, 1364, 1275, 1180 cm-1.1Н NMR (500 MHz, CDCl3) δ = 7.39-8.04 (m, 10H, 2 x СОC6H5), 6.21 (d, 1Н, J1,2 =5.7 Hz, Н-1), 5.59 (d, 1Н, J3,4 = 3.2 Hz, Н-3), 4.86 (d, 1Н, J2,1 = 5.7 Hz, Н-2), 4.64 (d, 2Н, H-5, Н-5´), 4.19-4.25 (m, 1Н, Н-4), 2.1 (s, 3H, NCH3). 13C NMR (126 MHz, CDCl3) δ = 166.7 (CN), 166.1 and 165.2 (C=O, 2хСОC6H5), 140.2, 140.0, 131.3, 131.2, 128.9, 128.8, 128.7, 128.2 (2хСОC6H5), 100.8 (С-1), 84.6 (C-4), 76.3, 75.5 (C-2, С-3), 61.6 (С-5), 13.8 (NMe). HRMS (ESI+):m/z calcd for C21H19NO6 (M+H)+: 382.1285, found 382.1287.
2-Phenyl-(3,5-di-О-benzoyl-α-D-xylofurano)-(1,2-d)-2-oxazoline (10).
Yield(97%), a colorless oil (method a1). (α)D20-27.5 (c 1.0, CHCl3). IR (KBr): ν 1725, 1665, 1268, 1112 cm-1.1Н NMR (500 MHz, CDCl3) δ = 7.42-8.13 (m, 15H, 2 x СОC6H5, -N=C-C6H5), 6.52 (d, 1Н, J1,2 =4.0 Hz, Н-1), 5.79 (d, 1Н, J3,4 = 3.0 Hz, Н-3), 5.15 (d, 1Н, J2,1 = 4.0 Hz, Н-2), 4.74 (d, 2Н, H-5, Н-5´), 4.33-4.36 (m, 1Н, Н-4). 13C NMR (126 MHz, CDCl3) δ = 167.0 (CN), 166.1 and 165.4 (C=O, 2хСОC6H5), 133.9, 133.2, 132.8, 129.9, 129.8, 129.2, 128.7, 128.6, 128.4 (2хСОC6H5, -N=C-C6H5), 100.8 (С-1), 84.8 (C-4), 76.4, 75.6 (C-2, С-3), 61.5 (С-5). HRMS (ESI+):m/z calcd for C26H21NO6 (M+Na)+: 466.1261, found 466.1263.
2-Ethyl-(3,5-di-О-benzoyl-α-D-xylofurano)-(1,2-d)-2-oxazoline (11).
Yield (86%), foam,(method a1). (α)D20- 36.3 (c 1.0, CHCl3). IR (KBr): ν 1724, 1670, 1363, 1275, 1182 cm-1. 1Н NMR (500 MHz, CDCl3) δ = 7.44-8.09 (m, 10H, 2 x СОC6H5), 6.29 (d, 1Н, J1,2 = 5.7 Hz, Н-1), 5.65 (d, 1Н, J3,4 = 3.2 Hz, Н-3), 4.92 (d, 1Н, J2,1 = 5.7 Hz, Н-2), 4.70 (d, 2Н, H-5, Н-5´), 4.21-4.28 (m, 1Н, Н-4), 2.45-2.49 (m, 2Н, -N=C-СН2СН3), 1.29 (t, 3H, -N=C-СН2СН3). 13C NMR (126 MHz, CDCl3) δ = 173.1 (CN), 166.2 and 165.3 (C=O, 2хСОC6H5), 133.8, 133.3, 129.9, 129.8, 129.5, 128.9, 128.7, 128.4 (2хСОC6H5), 100.6 (С-1), 84.5 (C-4), 76.3, 75.4 (C-2, С-3), 61.6 (С-5), 21.5 (N=C-СН2СН3), 10.2 (N=C-СН2СН3). HRMS (ESI+): m/z calcd for C21H21NO6 (M+H)+: 396.1442, found 396.1446.
2-Methyl-(3,5-О-isopropylidene-α-D-xylofurano)-(1,2-d)-2-oxazoline (12).
Yield (76%), oil (methoda1).(α)D20+6.9 (c 1.0, CHCl3). IR (film): ν 2993, 2940, 1669, 1384, 1228, 1043 cm-1. 1Н NMR (500 MHz, CDCl3) δ = 6.10 (d, 1Н, J1,2 = 5.5 Hz, Н-1), 4.65 (d, 1Н, J2,1 = 5,5 Hz, Н-2), 4.30 (d, 1Н, J3,4 = 2.4 Hz,Н-3), 4.09 (dd, 1Н, J5,4 = 2.5, J5,5′ = 13.6 Hz, Н-5), 4,07 (d, 1Н, Н-5′), 3.47-3.3.49 (m, 1Н, Н-4), 2.00 (s, 3H, NCH3), 1.42 and 1.36 (2 s, 3H, (CH3)2C-)). 13C NMR (126 MHz, CDCl3) δ: 168.3 (CN), 101.3 (С-1), 97.7 ( C-CH3)2), 85.9 (C-4), 72.9, 69.6 (C-2, С-3), 59.5 (С-5), 28.8 and 18.6 ((CH3)2C-), 13.7 (NMe). HRMS (ESI+): m/z calcd for C10H15NO4 (M+H)+: 214.1074, found 214.1084.
2-Methyl-(3,5-di-О-benzoyl-α-D-ribofurano)-(1,2-d)-2-oxazoline (13).
Yield (99%), a colorless oil (method a1). (α)D20+74.5 (c 1.0, CHCl3). IR (film): ν 1729, 1665, 1272, 1119 cm-1. 1Н NMR (500 MHz, CDCl3) δ = 7.40-8.08 (m, 10H, 2 x СОC6H5), 6.17 (d, 1Н, J1,2 =5.5 Hz, Н-1), 5.22 (t, 1Н, J2,1 =5.5, J2,3 =5.7 Hz, Н-2), 5.16 (dd, 1Н, J3,4 = 9.0 Hz, Н-3), 4.75 (dd, 1Н, J5,4 =3.6, J5,5′ =12.0 Hz, Н-5), 4.57 (dd, 1Н, J5′,4 =5.2 Hz, Н-5′), 4.19-4.23 (m, 1Н, Н-4), 2.1 (s, 3H, NCH3). 13C NMR (CDCl3) δ = 169.7 (CN), 166.1 and 165.5 (C=O, 2хСОC6H5), 133.6, 133.2, 129.8, 129.7, 129.4, 128.8, 128.5, 128.3 (2хСОC6H5), 100.6 (С-1), 78.5 (C-4), 74.1, 74.0 (C-2, С-3), 63.0 (С-5), 13.8 (NMe). HRMS (ESI+):m/z calcd for C21H19NO6 (M+H)+: 382.1285, found 382.1287.
2-Phenyl-(3,5-di-О-benzoyl-α-D-ribofurano)-(1,2-d)-2-oxazoline (14).
Yield (97%), a white solid (method a1). M.p. 128-129 oC. (α)D20+113.8 (c 1.0, CHCl3). IR (KBr): ν 1739, 1716, 1646, 1277, 1101 cm-1.1Н NMR (500 MHz, CDCl3) δ = 7.35-8.04 (m, 15H, 2 x СОC6H5, -N=C-C6H5), 6.37 (d, 1Н, J1,2 =5.6 Hz, Н-1), 5.38 (t, 1Н, J3,2 = 5.7 Hz, Н-2), 5.24 (d, 1Н, J3,4 =J3,2 = 5.9 Hz, Н-3), 4.72 (dd, 1Н, J5,4 = 4.7, J5,5́ = 12.1 Hz, H-5), 4.55 (dd, 1Н, J5´,4 = 4.7 Hz, Н-5´), 4.20-4.24 (m, 1Н, Н-4). 13C NMR (126 MHz, CDCl3) δ = 167.5 (CN), 166.1 and 165.7 (C=O, 2хСОC6H5), 133.6, 133.1, 132.5, 129.8, 129.7, 129.4, 128.9, 128.8, 128.5 (2хСОC6H5, -N=C-C6H5), 100.8 (С-1), 78.4 (C-4), 74.1, 74.09 (C-2, С-3), 63.0 (С-5). HRMS (ESI+):m/z calcd for C26H21NO6 (M+Na)+: 466.1261, found 466.1264.
2-Methyl-(3,5-di-О-benzoyl-β-D-arabinofurano)-(1,2-d)-2-oxazoline(16).
Yield (99%), a colorless oil (methoda1).(α)D20-36.1 (c 1.0, CHCl3). IR (KBr): ν 1725, 1669, 1321, 1269, 1108 см-1.1Н NMR (500 MHz, CDCl3) δ = 7.41-8.06 (m, 10H, 2 x СОC6H5), 6.14 (d, 1Н, J1,2 = 5.4 Hz, Н-1), 5.51 (br.d, 1Н, J3,4 = 2.5, J3,2 = 1.2 Hz, Н-3), 4.98 (br. d, 1Н, J2,1 = 5.5 Hz, Н-2), 4,54-4,57 (m, 1Н, Н-4), 4,39 (dd, 1Н, J5,4 = 6.2, J5,5′ =11.7 Hz, Н-5), 4.36 (dd, 1Н, J5′,4=3.9 Hz, Н-5′), 2.07 (s, 3H, NCH3). 13C NMR (126 MHz, CDCl3) δ = 168.7 (CN), 166.1 and 165.4 (C=O, 2хСОC6H5), 133.7, 133.1, 129.8, 129.8, 129.6, 128.8, 128.5, 128.3 (2хСОC6H5), 101.8 (С-1), 86.4 (C-4), 80.7, 79.0 (C-2, С-3), 63.7 (С-5), 14.3 (NMe). HRMS (ESI+):m/z calcd for C21H19NO6 (M+H)+: 382.1285, found 382.1287.
2-Phenyl-(3,5-di-О-benzoyl-β-D-arabinofurano)-(1,2-d)-2-oxazoline (17).
Yield (97%), a colorless oil (methoda1).(α)D20-8.5 (c 1.0, CHCl3). IR (film): ν 1721, 1642, 1269, 1109 cm-1. 1Н NMR (500 MHz, CDCl3) δ = 7.30-8.14 (m, 15H, 2 x СОC6H5, -N=C-C6H5), 6.46 (d, 1Н, J1,2 =5.7 Hz, Н-1), 5.76 (d, 1Н, J3,4 = 2.6 Hz, Н-3), 5.29 (d, 1Н, J2,1 = 5.7 Hz, Н-2), 4.65-4.68 (m, 1Н, Н-4), 4.48 (dd, 1Н, J5,4 5.8, J5,5′ = 11.6 Hz, Н-5), 4.38 (dd, 1Н, J5′,4 = 6.4 Hz, Н-5′). 13C NMR (126 MHz, CDCl3) δ = 166.9 (CN), 166.1 and 165.5 (C=O, 2хСОC6H5), 133.8, 133.1, 132.8, 129.9, 129.8, 129.2, 128.7, 128.6, 128.3 (2хСОC6H5, -N=C-C6H5), 100.9 (С-1), 86.7 (C-4), 81.4, 79.3 (C-2, С-3), 63.8 (С-5). HRMS (ESI+): m/z calcd for C26H21NO6 (M+Na)+: 466.1261, found 466.1265.
2-Methyl-(3,5,6-tri-О-benzoyl-α-D-glucofurano)-(1,2-d)-2-oxazoline (19).
Yield (93%), a colorless oil (methoda1).(α)D20-173.0 (c 1.0, CHCl3). IR (film): ν 1725, 1667, 1375, 1266 cm-1. 1Н NMR (500 MHz, CDCl3) δ = 7.40-8.04 (m, 15H, 3 x СОC6H5), 6.26 (d, 1Н, J1,2 = 5.6 Hz, Н-1), 5.90-5.93 (m, 1H, Н-5), 5.62 (dd, 1Н, J3,4 = 3.1 Hz, Н-3), 5.02 (dd, 1Н, Н-6), 4.90 (d, 1Н,Н-2), 4.63 (dd, 1Н, Н-6′), 4.27 (dd, 1Н, Н-4), 2.18 (s, 3H, NCH3). 13C NMR (125 MHz, CDCl3) δ = 169.04 (CN), 166.11, 165.3 and 165.1 (C=O, 3хСОC6H5), 133.69, 133.27, 133.06, 131.18, 129.9, 129.73, 129.68, 128.56 128.37 (3хСОC6H5), 100.88 (С-1), 84.56, 75.71, 75.64, 75.51 (C-4, C-5, C-2, С-3), 64.25 (С-6), 13.9 (NMe). HRMS (ESI+):m/z calcd for C29H25NO8 (M+Na)+: 538.1478, found 538.1453.
2-Phenyl-(3,5,6-tri-О-benzoyl-α-D-glucofurano)-(1,2-d)-2-oxazoline (20).
Yield (92%), a colorless oil (methoda1).(α)D20-34.6 (c 1.0, CHCl3). IR (KBr): ν 1728, 1646, 1268, 1102 cm-1.1Н NMR (500 MHz, CDCl3) δ = 7.36-8.15 (m, 20H, 2 x СОC6H5, -N=C-C6H5), 6.52 (d, 1Н, J1,2 = 5.5 Hz, Н-1), 5.95-5.99 (m, 1Н, Н-5), 5.75 (d, 1Н, J3,4 = 3.0 Hz, Н-3), 5.13 (d, 1Н, J2,1 = 5.5 Hz, Н-2), 5.06 (dd, 1Н, J6,5 = 5.8, J6,6′ = 11.6 Hz, H-6), 4.65 (dd, 1Н, J6,5 = 5.4 Hz, H-6´), 4.36 (dd, 1Н, Н-4). 13C NMR (126 MHz, CDCl3) δ = 166.9 (CN), 166.0, 165.3 and 165.0 (C=O, 3хСОC6H5), 133.6, 133.1, 132.9, 132.7, 129.8, 129.6, 128.6, 128.5, 128.2 (3хСОC6H5, -N=C-C6H5), 100.9 (С-1), 84.8, 75.6, 75.6, 68.4 (C-5, C-4, C-2, С-3), 64.2 (С-6). HRMS (ESI+):m/z calcd for C34H27NO8 (M+Na)+: 600.1634, found 600.1630.
2-Methyl-(3,5,6-tri-О-acetyl-α-D-glucofurano)-(1,2-d)-2-oxazoline (22).
Yield (95%), a colorless oil (methoda1).(α)D20+7.2 (c 1.0, CHCl3). IR (film): ν 1743, 1662, 1375, 1243 cm-1. 1Н NMR (500 MHz, CDCl3) δ = 6.07 (d, 1Н, J1,2=5.6 Hz, Н-1), 5.34 (br.s, 1H, H-3), 5.23-5.26 (m, 1H, Н-5), 4.63 (d, 1Н, J3,4 = 3.1 Hz, Н-2), 4.54(dm, 1H, H-4), 4.04 (dd, 1Н, Н-6), 3.82 (dd, 1Н,Н-6’), 2.06, 2.05, 2.02, and 1.97 (4s, 3H, 3x COCH3, NCH3).13C NMR (126 MHz, CDCl3) δ= 170.6 (CN), 169.6, 169.6 and 168.8 (C=O, 3хСОCH3), 100.9 (С-1), 84.5, 75.2, 74.3, 67.5 (C-4, C-5, C-2, С-3), 63.4 (С-6), 20.8 and 20.7 (3хСОCH3), 13.9 (NMe). HRMS (ESI+): m/z calcd for C14H19NO8 (M+Na)+: 352.1003, found 352.1006.
2-Phenyl-(3,5,6-tri-О-acetyl-α-D-glucofurano)-(1,2-d)-2-oxazoline (23).
Yield (92%), a white solid (method a1).M.p. 169-172 0С.(α)D20+25.0 (c 1.0, CHCl3).1Н NMR (500 MHz, CDCl3) δ = 7.43-8.02 (m, 5H, Ph), 6.33 (d, 1Н, J1,2=5.5 Hz, Н-1), 5.49 (d, 1H, J3,2 = 3.0 Hz, H-3), 5.31 (ddd, 1H, Н-5), 4.86 (d, 1Н, J2,1 = 5.5 Hz, Н-2), 4.60 (dd, 1Н, J6,5 = 2.1, J6,6′ = 12.3 Hz, Н-6), 4.08 (dd, 1Н, J6,5 = 5.8, Н-6'), 3.91 (dd, 1H, H-4), 2.11, 2.01 and 1.99 (3s, 3H, 3xCOCH3).13C NMR (126 MHz, CDCl3) δ: 170.7 (-C=N), 169.75, 169.71 and 166.6 (C=O, 3хСОCH3), 113.3, 129.1, 128.6, 115.9 (Ph-C=N-), 101.1 (С-1), 84.5, 75.4, 74.5, 67.5 (C-4, C-5, C-2, С-3), 63.4 (С-6), 20.81 and 20.76 (3хСОCH3). HRMS (ESI+): m/z calcd for C18H21NO8 (M+Na)+: 402.1003, found 402.1008.
2-Phenyl-(3,5,6-tri-О-benzoyl-α-D-allofurano)-(1,2-d)-2-oxazoline (25).
Yield (86%), a colorless oil (method a1).(α)D20+58.5 (c 1.0, CHCl3). IR (film): ν 1728, 1649, 1265, 1118 cm-1.1Н NMR (500 MHz, CDCl3) δ = 7.29-8.00 (m, 15H, 3 x СОC6H5), 6.40 (d, 1Н, J1,2 = 5.6 Hz, Н-1), 5.87-5.90 (m, 1H, Н-5), 5.43-5.47 (m, 2Н,H-2 and Н-3), 4.88 (dd, 1Н, J6,5 = 3.3, J6,6′ = 12.1 Hz, Н-6), 4.68 (dd, 1Н, J6,5 = 6.8 Hz, Н-6′), 4.27 (dd, 1Н, Н-4). 13C NMR (125 MHz, CDCl3) δ = 166.8 (-C=N), 166.1, 165.54 and 165.5 (C=O, 3хСОC6H5, C6H5), 133.4, 133.2, 133.1, 132.6, 129.8, 129.7, 129.0, 128.5, 128.4, 128.3,128.2 (3хСОC6H5, Ph-C=N-), 100.8 (С-1), 78.6, 75.1, 74.7, 71.2 (C-4, C-5, C-2, С-3), 63.4 (С-6). HRMS (ESI+):m/z calcd for C34H27NO8 (M+H)+: 578.1810, found 578.1816; (M+Na)+: 600.1634, found 600.1637.
2-Methyl-(3,5,6-tri-О-acetyl-α-D-allofurano)-(1,2-d)-2-oxazoline (27).
Yield (98%), a colorless oil (methoda1).(α)D20+107.2 (c 1.0, CHCl3). IR (film): ν 17440, 1667, 1375, 1247 cm-1. 1Н NMR (500 MHz, CDCl3) δ = 6.01 (d, 1Н, J1,2=5.6 Hz, Н-1), 5.34 (dm, 1H, H-5), 5.02 (t, 1Н, J2,3 = 5.8 Hz, Н-2), 4.94 (dd, 1H, H-3), 4.41(dd, 1H, H-4), 4.14 (dd, 1Н, Н-6), 3.80 (dd, 1Н,Н-6’), 2.16, 2.11, 2.10, and 2.08 (4s, 3H, 3x COCH3, NCH3). 13C NMR (126 MHz, CDCl3) δ = 170.5 (CN), 169.8, 169.7 and 169.6 (C=O, 3хСОCH3), 100.4 (С-1), 78.2, 74.5, 73.9, 70.0 (C-4, C-5, C-2, С-3), 62.3 (С-6), 20.8, 20.7 and 20.4(3хСОCH3), 13.9 (NMe). HRMS (ESI+): m/z calcd for C14H19NO8 (M+Na)+: 352.1003, found 352.1004.
Synthesis of 2-O-isopropyl-3-O-p-toluenesulfonyl-5-O-benzoylxylitol (30).
To a stirred solution of acetonide 3 (147 mg, 0.33 mmol) in anhydrous acetonitrile (1.9 ml) KHF2 (103 mg, 1.32 mmol), NaBH4 (59 mg, 1.5 mmol) and then boron trifluoride diethyl etherate (1.42 ml, 11.38 mmol) were added successively. The reaction mixture was stirred at room temperature for 18 h, and then was gradually poured into cooled 5% NaHCO3.The aqueous phase was extracted with CHCl3 (3x30 ml). The combined organic extracts were washed water, dried over anh. Na2SO4, and evaporated to dryness. The residue was chromatographed on silica gel, using for elution a mixture of hexane-ethylacetate 6:1 and 3:1, and 1:2 as the eluent to give the starting acetonide 3 (38 mg, 26%) and the xylitol derivative 30 (59 mg, 40%) as a colorless oil. (α)D20+1.4 (c 0.5, CHCl3). 1Н NMR (500 MHz, CDCl3) δ = 7.30-8.03 (m, 9H, СОC6H5 and ОSO2C6H4CH3), 4.84 (dd, 1Н), 4.31-4.38 (m, 2Н), 3.90-3.97 (m, 3Н), 3.82-3.91 (m, 2Н), 2.41 (s, 3H, ОSO2C6H4CH3), 1.22 (d, 3H, (CH3)CH-), 1.21 (d, 3H, (CH3)CH-). 13C NMR (126 MHz, CDCl3) δ = 165.9 (C=O, СОC6H5), 145.4, 133.3, 130.0, 129.8, 129.7, 128.5 (СОC6H5 andОSO2C6H4CH3), 78.3, 75.5, 72.2, 65.5, 64.5 (-CH2OBz), 59.9 (-CH2OH), 22.8 ((CH3)2CH-), 22.7 ((CH3)2CH-), 21.7 (ОSO2C6H4CH3). HRMS (ESI+): m/z calcd for C22H22O8SNa (M+Na)+: 475.1403, found 475.1391.
Synthesis of 2-O-isopropyl-3-O-p-toluenesulfonyl-1,4,5-tri-O-benzoylxylitol (31).
To a stirred solution of xylitol derivative 30 (45 mg, 0.099 mmol) in anhydrous pyridine (2 ml) BzCl (0.068 ml, 0.57 mmol) was added at 0 oС and then the reaction mixture was stirred for 48 h at room temperature, diluted with CH2Cl2 and poured into cold 5% aq NaHCO3. The aqueous phase was extracted with CH2Cl2 (3x50 ml), the combined organic extracts were washed with water, dried and evaporated. The residue was chromatographed on a silica gel, using for elution a mixture of hexane-ethylacetate 6:1, 5:1, and chloroform to give (42.7 mg,65%) of protected xylitol derivative 31 as a colorless oil. (α)D20-17.8 (c 0.79, CHCl3).1Н NMR (500 MHz, CDCl3) δ = 7.40-8.18 (m, 15H, 3xСОC6H5), 7.77 and 7.11 (2d, 4H, ОSO2C6H4CH3), 5.91 (q, 1Н, H-4), 5.26 (dd, 1H, H-3 ), 4.64 (dd,1H, H-5), 4.60 (dd,1H, H-5′), 4.54 (dd, 1H, H-1), 4.47 (dd, 1H, H-1′), 4.19-4.22 (m, 1Н, H-2), 3.89-3.94 (m, 1Н, (CH3)2CH-), 2.30 (s, 3H, ОSO2C6H4CH3), 1.25 (d, 3H, (CH3)CH-), 1.21 (d, 3H, (CH3)CH-). 13C NMR (126 MHz, CDCl3) δ = 166.1, 165.8, and 165.3 (C=O, 3xСОC6H5), 145.0, 133.8, 133.4, 133.2, 133.16, 130.2, 130.0, 129.8 (СОC6H5 andОSO2C6H4CH3), 77.9, 73.5, 72.4, 69.6, 62.7, 62.6, 22.9 ((CH3)2CH-), 22.2 ((CH3)2CH-), 21.6 (ОSO2C6H4CH3). HRMS (ESI+): m/z calcd for C36H36O10SNa (M+Na)+: 683.1927, found 683.1929.
Synthesis of 2-methyl-(5-О-benzoyl-α-D-xylofurano)-(1,2-d)-2-oxazoline (49) from 5-O-benzoyl-D-xylofuranose (47).
b1. To a stirred solution of 5-O-benzoyl xylofuranose 47 (49 mg, 0.19 mmol) in anhydrous acetonitrile (2.5 ml) KHF2 (57 mg, 0.89 mol) and boron trifluoride diethyl etherate (0.14 ml, 1.10 mmol) were added successively. The reaction mixture was stirred at room temperature for 3 h, and then poured into cooled 5% aq NaHCO3. The aqueous phase was extracted with CHCl3 (3x50 ml). The combined organic extracts were washed with water, dried over anh. Na2SO4, and evaporated to dryness. The oxazoline 49 (40 mg, 75%) was prepared as a colorless oil. (α)D20-15.9 (c 0.56, CHCl3). M.p. 63-65 oC (crystallized under storing).1Н NMR (500 MHz, CDCl3) δ = 7.49-8.09 (m, 5H, СОC6H5), 6.14 (d, 1Н, J1,2 = 5.5 Hz, Н-1), 4.86 (dd, 1Н, J5,4 = 7.4 Hz, J5,5′ = 11.6 Hz, Н-5), 4.82 (d, 1Н, J2,1 = 5.6 Hz, Н-2), 4.47 (dd, 1Н, J5′,4 = 5.1 Hz, Н-5′), 4.27 (d, 1Н, J3,4 = 2.3 Hz, Н-3), 3.86-3.89 (m, 1Н, Н-4), 2.09 (s, 3H, NCH3). 13C NMR (126 MHz, CDCl3) δ = 168.9 (CN), 167.3 (C=O, СОC6H5), 100.0 (С-1), 86.6 (C-4), 76.7, 74.1 (C-2, С-3), 61.3 (С-5), 13.9 (NMe).HRMS (ESI+): m/z calcd for C14H15NO5 (M+H)+: 278.1028, found 278.1025.
Synthesis of 2-methyl-(3,5-di-О-benzoyl-α-D-ribofurano)-(1,2-d)-2-oxazoline (13) from 1,3,5-tri-O-benzoyl-α-D-ribofuranose (50).
b2. To a stirred solution of 1,3,5-tri-O-benzoyl-α-D-ribofuranose (50) (250 mg, 0.54 mmol) in anhydrous acetonitrile (10.0 ml) KHF2 (156 mg, 1.99 mmol) and boron trifluoride diethyl etherate (0.39 ml, 3.08 mmol) were added successively. The reaction mixture solution was stirred at room temperature for 3.5 h, and then poured into cooled 1N aq NaOH. The oxazoline 13 (204 mg, 99%) was prepared as a colorless oil after the work-up.
c2. To a stirred solution of ribofuranose derivative 50 (100 mg, 0.22 mmol) in anhydrous acetonitrile (4.0 ml) boron trifluoride diethyl etherate (0.16 ml, 1.26 mmol) was added. The reaction mixture solution was stirred at room temperature for 3.5 h, and then poured into cooled 2.7 ml 1N aq NaOH. The oxazoline 13 (90 mg) as yellowish oil was prepared in 65% yield estimated by 1H NMR in CDCl3.
2-Phenyl-(β-D-arabinofurano)-(1,2-d)-2-oxazoline (51).
3,5-Di-O-benzoyl oxazoline derivative 17 (95 mg, 0.21 mmol) was dissolved in 7 ml methanol saturated at 0 oC with ammonia, then reaction mixture was left for 14 h at room temperature and evaporated to dryness. The residue was chromatographed on a silica gel, using for elution chloroform, chloroform-methanol 15:1, 10:1 and 6:1 to give (47 mg, 93%) of the oxazoline 51 as oil. (α)D20-18.7 (c 0.56, MeOH). 1Н NMR (500 MHz, CD3OD) δ = 7.99-7.49 (m, 5H, -N=C-C6H5), 6.18 (d, 1Н, J1,2 = 6.2 Hz, Н-1), 5.03 (dd, 1Н, J2,3 = 1.3 Hz, Н-2), 4.36 (br.d, 1Н,Н-3), 3.98-4.01 (m, 1Н, Н-4), 3.49 (dd, 1Н, J5,4 = 6.0, J5,5′ = 11.8 Hz, Н-5), 3.44 (dd, 1Н, J5′,4 = 6.1 Hz, Н-5′). 13C NMR (126 MHz, CD3OD) δ = 168.74 (CN), 134.08, 130.01 and 128.04 (N-C6H5), 101.98 (С-1), 90.85 (C-4), 87.31, 77.86 (C-2, С-3), 62.93 (С-5). HRMS (ESI+): m/z calcd for C12H13NO4 (M+H)+: 236.0923, found 236.0927.
2-Phenyl-(3,5-di-O-acetyl β-d-arabinofurano)-(1,2-d)-2-oxazoline (52).
The oxazoline 51 (20 mg, 0.085 mmol) was dissolved in 1.7 ml anhydrous pyridine, acetic anhydride (0.04 ml, 0.42 mmol) was added, then reaction mixture was stirred for 48 h at room temperature and then poured into a mixture of ice and water. The aqueous phase was extracted with CH2Cl2 (3x20 ml). The combined organic extracts were washed with water, dried over anh. Na2SO4, and evaporated to dryness. The residue was chromatographed on a silica gel, using for elution elution mixtures of ethylacetate-petroleum ether to give (22 mg, 80%) of the oxazoline 52 as oil. (α)D20 -4.6 (c 0.3, CHCl3). 1Н NMR (500 MHz, CDCl3) δ = 7.42-8.03 (m, 5H, -N=C-C6H5), 6.30 (d, 1Н, J1,2 = 6.2 Hz, Н-1), 5.32 (br.d, 1Н,Н-3), 5.04 (br.d, 1Н, J2,3 = 1.3, J2,1 = 6.2 Hz, Н-2), 4.26-4.33 (m, 1Н, Н-4), 4.06 (dd, 1Н, J5,4 = 5.9, J5,5′ = 11.8 Hz, Н-5), 4.02 (dd, 1Н, J5′,4 = 6.1 Hz, Н-5′), 2.15 and 1.89 (2s, 3H, -COCH3). 13C NMR (126 MHz, CD3OD) δ= 170.6, 169.9 and166.5 (2-COCH3 and CN), 132.7, 129.1, 128.8, 128.6 (N-C6H5), 102.2 (С-1), 86.1 (C-4), 81.2, 78.8 (C-2, С-3), 62.5 (С-5), 20.9 and 20.6 (2xCOCH3). HRMS (ESI+): m/z calcd for C16H17NO6 (M+Na)+: 3430940, found 343.0945.
Biological assays of antiproliferative activity
Cell culturing
Anti-proliferative activities of newly synthesized compounds were tested against myelogenous leukemia (K562), cervical carcinoma (Hela), breast carcinoma (MCF-7) in comparison with 5-fluorouracil as the positive control. Human cell lines were obtained from the Institute of Cytology, Russian Academy of Sciences. Human cell lines were cultured as monolayers and maintained in Eagle’s medium (DMEM) supplemented with 10% foetal bovine serum (FBS), 2 mM L-glutamine, 100 U/ml amoxicillin, 100 ϻg/ml streptomysin in a humidified atmosphere of 5% CO2 at 37 0 C. Stock solutions of compounds were prepared in DMSO and kept at – 200C. Controls were added with the final concentration of DMSO (0.01%).
Proliferation assays
The cytotoxic effects on human cancer cells were assessed after 72 h incubation of sugar oxazoline derivative in concentrations 0.1 - 50 ϻM with the cell culture in a 96-well flat-bottomed plate at 37 0C under conditions of 5% CO2 and 95% air humidity using resazurin assay with triplicate experiments. Aliquots of resazurin solution (10 ϻL) was added to each well and incubated for 3 h at 37 0C. In all experiments, DMSO controls were included. Fluorescene resorufin measurements were performed on a multimodal absorbance fluorimeter Infinite® 200 PRO (Tecan, Switzerland) at an excitation wavelength of 530 nm and an emission wavelength of 590 nm. IC50 values for each compound were calculated from the cell growth inhibition curves obtained from the treatments done with increasing concentrations.
References
- 1.F M Cordero, Lascialfori L, Machetti F. (2021) Five-membered ring systems with. , O and N atoms.InProgress,inHeterocycl. Chem. Gribble G.W., Joule J.A. Eds.; Elsevier.; 33, 311-340.
- 2.T G Gant, MeyersA I. (1994) . The CHEMISTRY of 2-OXAZOLINES (1985-PRESENT),Tetrahedron.50 2297-2360.
- 3.A J Fairbanks. (2018) Synthetic and semi-synthetic approaches to unprotected N-glycan oxazolines.Beilstein. , J. Org. Chem.14,416-429 https://doi.org, 10-3762.
- 4.T B Parsons, M K Patel, D J Vocadlo, A B Boraston, A J Fairbanks. (2010) Streptocococcus pneumoniae endohexosaminidase D: feasibility of using N-glycan oxazoline donors for synthetic glycosylation of a GlcNAc-asparagine acceptor,Org.Biomol.Chem. 8, 1861-1868.
- 5.Koda Y, Terashima T, Ouchi M. (2019) Unnatural Oligoaminosaccharides with N-1,2-Glycosidic Bonds Prepared by Cationic Ring-Opening Polymerization of 2-Oxazoline-Based Heterobicyclic. , Sugar Monomers,ACS Macro Lett 8, 1456-460.
- 6.Damkaci F, DeShong P. (2003) Stereoselective Synthesis of α- and β-Glycosamide Derivatives from Glycopyranosyl Azides via Isooxazoline Intermediates,J. , Am. Chem. Soc 125, 4408-4409.
- 7.Blanco J L J, E M Rubio, C O Mellet, Fernandez J M G. (2004) . Synthesis of Sugar Oxazolines by Intramolecular Ritter-Like Reaction of D-Fructose Precursors,Synlett.12 2230-2232.
- 8.Vangala M, G P Shinde. (2015) Synthesis of D-fructose-derived spirocyclic 2-substituted-2-oxazoline ribosides,Beilstein. , J. Org. Chem 11, 2289-2296.
- 9.E M Reid, E S Vigneau, S, C H Mazzabadi, H De Castra. (2012) One-Pot Synthesis of N-Glycoozazolines, N-Glycoaminooxazolines, and N-Glycothiazolines from Glycals.Eur. , Org. Chem 17, 3295-3303.
- 10.Andreini M, Anderluth M, Audfray A, Bernardi A, Imberty A. (2010) Monovalent and bivalent N-fucosyl amides as high affinity ligands for Pseudomonas aeruginosa PA-IIL lectin.Carbohydr. , Res 345, 1400-1407.
- 11.D M Gordon, S J Danishefsky. (1991) . Ritter-like Reactions of 1,2-Anhydropyranose Derivatives,J. Org. Chem.56 3713-3715.
- 12.Blanco J L J, Sylla B, C O Mellet, Fernandez J M G. (2007) Synthesis of α- and β-Glycosyl Isothiocyanates via Oxazoline Intermediates,J. , Org. Chem 72, 4547-4555.
- 13.Glorgydeák Z, Hadady Z, Felfoldi N, Krakomperger A, Nagy V. (2004) Synthesis of N-(β-D-glucopyranosyl)- and N-(2-acetamido-2-deoxy-β-D-glucopyranosyl) amides as inhibitors of glycogen phosphorylase,Bioorg. , Med. Chem 12, 4861-4820.
- 14.Czifrák K, Hadady Z, Docsa T, Gergely P, Schmidt J. (2006) Synthesis of N-(β-D-glucopyranosyl) monoamides of dicarboxylic acids as potential inhibitors of glycogen phosphorylase,Carbohydr. , Res 341, 947-956.
- 15.N E Poopeiko, E I Kvasuyk, I A Mikhailopulo, M J Lidak. (1985) Stereospecific Synthesis of β-D-Xylofuranosides of Adenine and Guanine,Synthesis. 6, 605-609.
- 16.G, A V Sivets. (2021) Synthesis of N-pentofuranosyl oxazolines and amides via selective transformations of acetonides of. D-sugars,Dokladyof the National Academy of Sciences of Belarus, ser. chem. Belarus ser. chem 65, 558-567.
- 17.Gosselin G, Puech F, Genu-Dellac C, Imbach J-L. (1993) 1,2-Di-O-acetyl-5-O-benzoyl-3-deoxy-3-fluoro-D-xylofuranose. A versatile precursor for the synthesis of 3-deoxy-3-fluoro-D-xylofuranosyl nucleosides as antiviral agents,Carbohydr.Res. 249, 1-17.
- 18.Roy V, Zerrouki R, Krauz P, Laumond G, A M. (2007) Synthesis and Antiviral Evaluation of Azt Analogues with A Spacer Arm, between Glucidic and Base Moieties. Part II,Nucleosides. , Nucleotides & Nucleic Acids.26 413-421.
- 19.Chen S-H, Lin S, King I, Spinka T, G E Dutschman. (1998) Synthesis and comparative evaluation of two antiviral agents: β-L-Fd4C and β-D-Fd4C,Bioorg. , Med. Chem. Lett 8, 3245-3250.
- 20.Soares F F P, M J Silva, Doboszewski B. (2013) Deoxygenation at the C3 position of D- and L-arabinofuranose: stereospecific access to enantiomeric cordycepose derivatives,Carbohydr. , Res 380, 143-148.
- 21.Genu-Dellac C, Gosselin G, Imbach J-L. (1992) Preparation of new acylated derivatives of L-arabinofuranose and 2-deoxy-L-erythro-pentofuranose as precursors for the synthesis of L-pentofuranosyl nucleosides,Carbohydr. , Res 216, 249-255.
- 22.Lajsic S, Miljkovic D, Cetkovic G. (1992) An improved synthesis of D-amicetose,Carbohydr. , Res 233, 261-264.
- 23.Haga M, Takano M, Tejima S. (1972) 3-O-Methyl-D-allose and a facile route to 2-and 3-O-methyl D-riboses,Carbohydr. , Res 21, 440-446.
- 24.R L Whistler, L W Doner. (1970) D-Glucopyranosylation of cellulose acetate,Carbohydr. Res.15 391-395.
- 25.Jiang D, He T, Ma L, WangZ. (2014) Recent developments. in Ritter reaction,RSC Advances 4, 64936-64946.
- 26.Guerinot A, Reymond S, Cossy J. (2012) Ritter Reaction: Recent Catalytic Developments,Eur. https.//doi.org. 10.1002/ejoc.201101018 , J. Org. Chem 1.
- 27.Klemer A, M J Kohla. (1988) Eine Einfache Synthese von N-Acyl Glykosylaminen,Carbohydr. , Chem 7, 785-797.
- 28.S A, C A Kilner, A G Leach, W B Turnbull. (2009) Neighbouring group participation vs. addition to oxacarbenium ions: studies on the synthesis of mycobacterial oligosaccharides,Org.Biomol. , Chem 7, 4842-4852.
- 29.Ch-Sh Chao, Ch-Yu Lin, Mulani S, W-Ch Hung, K-K T Mong. (2011) Neighboring-Group Participation by C-2 Ether Functions in Glycosylations Directed by Nitrile Solvents,Chem. , Eur. J 17, 12193-12202.
- 30.R, Behrendt M, Toepfer A. (1990) Nitriles as Solvents. in Glycosyl Reactions: Highly Selective β-Glycosid Synthesis,Synlett 11, 694-696.
- 31.Braccini I, Derouet C, Esnault J, C Herv’e du Penhoat, Mallet J-M. (1993) Conformational analysis of nitrilium intermediates in glycosylation reactions,Carbohydr. , Res 246, 23-41.
- 32.Kafle A, Liu j.Cui L.,(2016) Controlling the stereoselectivity of glycosylation via solvent effects,Can. , J. Chem.94 11, 894-901.
- 33.A, Lassfolk R, F S Ekholm, Leino R, Crich D. (2020) . Mechanisms of Stereodirecting Participation and Ester Migration from Near and Far in Glycosylation and Related Reactions,Chem. Rev.120 7104-7151.
- 34.K.Dae-Hwan S.,(2011) Remote Participation of Protecting Groups at Remote Positions of Donors. in Glycosylations,Trends inGlycoscienceandGlycotechnology.23 53-66.
- 35.Wierenga W, H L Skulnick. (1981) Stereochemical control as a function of protecting-group in 2-deoxy-d-erythro-d-pentofuranosyl nucleosides,Carbohydr. , Res 80, 41-52.
- 36.Kumar A, Kumar V, R T Dere.. Schmidt R.R.,(2011) Glycoside Bond Formation via Acid-base Catalysis,Org. Lett.13 14, 3612-3615.
- 37.Nielsen M, Pedersen C M. (2018) Сatalytic Glycosylations in Oligosaccharide Synthesis,Chem. , Rev 118, 8285-8358.
- 38.Kumar A, Geng Y.. Schmidt R.R.,(2012) Silicon Fluorides for Acid-Base Catalysis in Glycosidations.Adv.Synth.Catal.354 1489-1499.
- 39.Smith J R L, Norman R O C, M R Stillings. (1975) Synthesis of Oxazolines from. , Epoxides,J. Chem. Soc. Perkin Trans I 13, 1200-1202.
- 40.Howell H G, Brodfuehrer P R, Brundidge S P, Bengini D A, Sapino C. (1988) Antiviral Nucleosides. A Stereospecific, Total Synthesis of 2´-Fluoro-2´-Deoxy-β-D-arabinofuranosyl Nucleosides,J. , Org. Chem 53, 85-88.
Cited by (1)
This article has been cited by 1 scholarly work according to:
Citing Articles:
Journal of New Developments in Chemistry (2025) Crossref OpenAlex Semantic Scholar