Lysozyme-Induced Degradation of Chitosan: The Characterisation of Degraded Chitosan Scaffolds
Abstract
Up till now, chitosan has confirmed its versatile application in skin, cartilage and bone tissue engineering, as well as in drug delivery applications. This study is focused on enzymatic degradation of porous chitosan structures usually designed for mentioned purposes. In vitro degradation was monitored during four weeks of incubation at physiological temperature and in two different media, phosphate buffer saline solution and water. The scaffolds were characterised before and after enzymatic degradation using scanning electron microscopy and infrared spectroscopy with Fourier transformations (FTIR). According to the gravimetric analysis, higher weight loss of chitosan scaffolds was observed in buffered medium with respect to the water. The results implied that the total weight loss obtained in buffer involves physical dissolution of chitosan and lysozyme cleavage of glycoside bond. Importantly, FTIR identification of chitosan scaffolds after enzymatic degradation indicated the absence of lysozyme activity in water, indicating that weight loss is a result of the chitosan dissolution. This finding greatly impacts design of degradation experiments and characterisation of degradation behaviour of chitosan-based materials utilised as implants or drug delivery systems.
Author Contributions
Academic Editor: Lin Ye, Cardiff China Medical Research Collaborative, Division of Cancer and Genetics, Cardiff University School of Medicine, Cardiff, CF14 4XN, UK, Email: [email protected]
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2017 Andrea Lončarević, et al.
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Introduction
One of the most important derivatives of chitin is chitosan, poly (D-glucosamine), a product of chitin deacetylation process. Chitosan is a linear polysaccharide, crystalline polymer insoluble at pH greater than 71. Chitosan has proven to be biodegradable, with antibacterial activity and hydrophilic, possessing functional groups (–OH and –NH2) for secondary bonds formation2. Moreover, chitosan possesses high biocompatibility, good miscibility with other polymers and good coating properties for bulk implants. Even during degradation, chitosan-oligomers are found to be bioactive. Besides, chitosan-oligomers can act as probiotics that positively change intestinal microflora balance, inhibit growth of harmful bacteria, promote good digestion and boost immune function3, 4. Chitosan is nontoxic and has been approved by FDA for would dressing application5.
In the pharmaceutical field, chitosan is used as a drug delivery system for oral, nasal, parenteral and transdermal applications, as well as implant for gene delivery. High chemical reactivity has also led to several chitosan-drug systems for treating tumours6, 7, 8. Moreover, it is used for encapsulation of living cells as an inner core of composite spheres9. In many cases, cross-linked chitosan membranes are used (mostly with glutaraldehyde and genipin)10, 11, 12.
Numerous studies are focused on the development of chitosan implants as porous materials for the treatment of tissue defects (including cartilage, skin and bones) or to accelerate the tissue regeneration. The tissue restoration is a complex process that involves the interactions between the cells, extracellular matrix components and signalling compounds. During the healing process, certain regulatory mechanisms associated with inflammation and immune responses are initiated. Biodegradable implants that accelerate tissue healing have to fulfil certain requirements such as biocompatibility, bioactivity, mechanical stability and appropriate biodegradability. Such implants have to degrade at a rate similar to new tissue formation. Moreover, mechanical stability of the implant integrated with newly-formed extracellular matrix should correspond to the mechanical performance of surrounding tissue.
The biodegradability of chitosan in living organisms depends on the degree of deacetylation and molecular weight. Chitosan can be degraded by enzymes that hydrolyse linkages between glucosamine–glucosamine, glucosamine–N-acetyl-glucosamine and N-acetyl-glucosamine–N-acetyl-glucosamine units. In human body, chitosan degrades due to the activity of lysozyme and bacterial enzymes present in the colon. Likewise, degradation of chitosan can be carried out by chitosanase, chitin deacetylase and β-N-acetylhexosaminidase13, 14. The wide pallet of chitosan properties makes it extensively studied in terms of physical, chemical and biological properties.
One of the important properties of chitosan-based material is in vitro and in vivo degradation behaviour. Numerous in vitro studies have investigated the influence of different incubation conditions, such as type of a buffer solution, the pH of incubation medium, type and concentration of enzyme, molecular weight, crystallinity and degree of deacetylation on degradation behaviour15, 16. Most commonly used techniques to characterise the degradation behaviour included gravimetric analysis, gel permeation chromatography, quantity of reducing sugar ends, microstructure imaging using electronic microscopy etc. The in vitro degradation studies can estimate the materials behaviour in vivo. However, due to the complexity of biological environment in the body, it is difficult to simulate them in vitro. Lysozyme has been found in various human body fluids including serum with concentration of 4 – 13 mg/L and tears with 100-fold higher concentration17. This implies that chitosan degradation behaviour strongly depends on the implantation site.
The purpose of this study was to investigate the influence of lysozyme on the degradation behaviour under different degradation conditions (phosphate buffer saline solution and water). The important results obtained by the FTIR identification technique indicated that lysozyme activity strongly depends on pH value of incubation medium which in the end determines the mechanism of chitosan weight loss.
Experimental Procedure
Materials
Chitosan (CHT) with deacetylation degree of 95 – 98 % and molecular weight, MW, of 100 000 – 300 000 g/mol was purchased from Across Organics (Belgium). Acetic acid (99.5 %) was purchased from Poch Gliwice (Poland), sodium hydroxide (NaOH) from Gram-Mol (Croatia), and ethanol (EtOH, 96 %) from Kefo (Slovenia). All chemicals were of analytical grade. Enzymatic degradation was performed with lysozyme (LZ, ≥ 90 % proteins, activity ≥ 40 000 U/mg, Sigma-Aldrich, Canada).
The molecular weight of chitosan was analysed using a gel permeation chromatographer at 35 °C using Waters Breeze GPC system with a 1525 Binary HPLC pump (Waters Corporation, Milford, MA) equipped with a 2414 refractive index detector and four serial columns (Ultra hydrogel 7.8 mm ID X 30 cm). The chitosan powder was dissolved in acetic buffer (0.5 mol/L CH3COOH/0.2 mol/L CH3COONa) with pH = 4.5 which was used as a mobile phase at a flow rate of 0.5 mL/min and 20 µL injection volume. The average molecular weight (Mw) of chitosan was determined to be 220 000.
Preparation of Chitosan Scaffold
The porous chitosan structures were obtained by thermally induced phase separation as described elsewhere18. Briefly, chitosan was dissolved in 0.36 % (w/w) acetic acid resulting in 1.2 % (w/v) chitosan solution. The solution was frozen in an aluminium mould at –22 °C over night. Then, frozen samples were immersed into neutralization medium consisting of 1 mol/L NaOH and EtOH (volume ratio 1:1) at –22 °C for 12 h. Afterwards, samples were immersed into EtOH at –22 °C for 12 h and dehydrated by immersion into ethanol at ambient temperature for next 24 h. Dehydrated samples were left to dry at atmospheric conditions.
Characterization of Chitosan Scaffold
The morphology of scaffolds was imaged using scanning electron microscope TESCAN Vega3SEM Easyprobe with electron beam energy of 10 keV. Previously to imaging, samples were sputtered with palladium/gold plasma for 120 s. The estimation of pore size distribution was determined on 280 pores measured on different chitosan samples.
The porosity of chitosan scaffolds (n = 3) was estimated using Archimedes principle performed in phosphate buffer saline solution (PBS, ρ = 1.0 g/mL) at ambient temperature. The open porosity (φ) of scaffold was calculated according to the following equation:
 (1)
(1)
Vporeis calculated as the weight of absorbed solution (PBS) divided by the density of the buffer. The amount of water absorbed in the pores was corrected by the swelling of non-porous chitosan samples. The scaffold’s volume (Vscaffold) was calculated as the ratio of scaffolds weight and experimentally determined density of non-porous chitosan film (ρ = 1.97 g/mL).
The swelling capacity was evaluated by scaffold’s immersion in phosphate buffer saline solution (pH = 7.5) and distilled water (pH = 6.50). Previously to immersion, scaffolds (n = 3) were weighted to obtain initial weight and subsequently immersed for 24 h at ambient temperature. Swollen samples were carefully weighted. The capacity of scaffold to absorb water was calculated as a difference in weight before and after swelling with respect to the initial weight of the sample.
The mechanical performance of chitosan scaffold was evaluated in terms of compressive and tensile strength in wet state (PBS, pH = 7.5) at ambient temperature. The compressive test was performed on previously swollen cylindrically-shaped samples (n = 5, diameter of 8 mm) with mechanical instrument Seiko TMA/SS6000 (Seiko Instrument Inc., Japan) with deformation rate of 50 µm/min. The tensile test was carried out by the same instrument on previously swollen samples (n = 5, diameter of 8 mm, height between 1 and 2 mm) until 10% of deformation with deformation rate of 1000 µm/min. The compressive and tensile moduli were determined as a slope of the stress–strain curve.
The scaffolds identification was carried out by infrared spectroscopy with Fourier transformations (FTIR) with a diamond crystal (Bruker Vertex 70) at 20 °C, in the spectral range of 4000 – 400 cm-1 with 24 scans and 4 cm-1 resolution.
In vitro Degradation Test
The degradation of chitosan scaffolds (n = 3) was performed in two media: phosphate buffer saline solution (PBS, pH = 7.4) and distilled water containing 800 mg/L of lysozyme at 37 °C in an orbital shaker at 50 rpm. Enzymatic degradation was monitored during four weeks, while the lysozyme solution was refreshed every third day. At predetermined time (1, 2, 3 and 4 weeks) samples were removed from the medium, rinsed with distilled water and dried in the oven at 50 °C until constant mass. The samples were weighted before (m1) and after in vitro degradation (m2). The degradation degree (Δm) was determined as the weight loss with respect to the initial weight of the sample:
 … (2)
… (2)
As a control, samples were incubated at the same conditions without lysozyme. During the degradation, pH of medium was measured by Schott CG 842 using BlueLine 14 electrode with a precision of 0.01.
Statistical analysis
Statistical comparison of the data was performed using ANOVA one-way test. A p value <0.05 was considered to be statistically significant. All data are presented as a mean value corrected by standard deviation (mean ± SD).
Results
Scaffold’s Microstructure
The microstructure of prepared chitosan scaffolds was investigated by SEM imaging. The highly porous structure is clearly observable in Figure 1a. The honeycomb-like pores with good interconnectivity were founded on scaffold’s cross section, which is important parameter in development of artificial grafts. Such microstructure allows a non-hindered diffusion of nutrients, oxygen and metabolic waste through entire volume of scaffold. The estimation of pore size using electronic microscopy (Figure 1b) indicates macroporous material with the majority of pores ranging from 100 to 250 µm.
Figure 1.SEM micrograph a); and pore size distribution b) of the cross section of chitosan scaffold.
Scaffold’s Physical Properties
Figure 2 summarises physical properties of chitosan scaffolds in terms of open porosity, swelling and mechanical performances under compressive and tensile loading. The estimated porosity of 98.5 ± 0.2% confirms highly porous structure observed by SEM analysis. Such porous structure allows material to absorb large quantity of water, as confirmed by the swelling ratio determined in PBS and water. The values of swelling ratio of 29.4 and 36.5 for PBS and water, respectively, indicate the ability of chitosan scaffold to absorb 30 times greater amount of water with respect to its weight. Apart from scaffold’s porosity, this ability originates also from the chitosan hydrogen nature.
Figure 2.Physical properties of chitosan scaffolds in terms of porosity, swelling and mechanical performances. Significant difference (p<0.05) between the groups is designated with asterisk (*).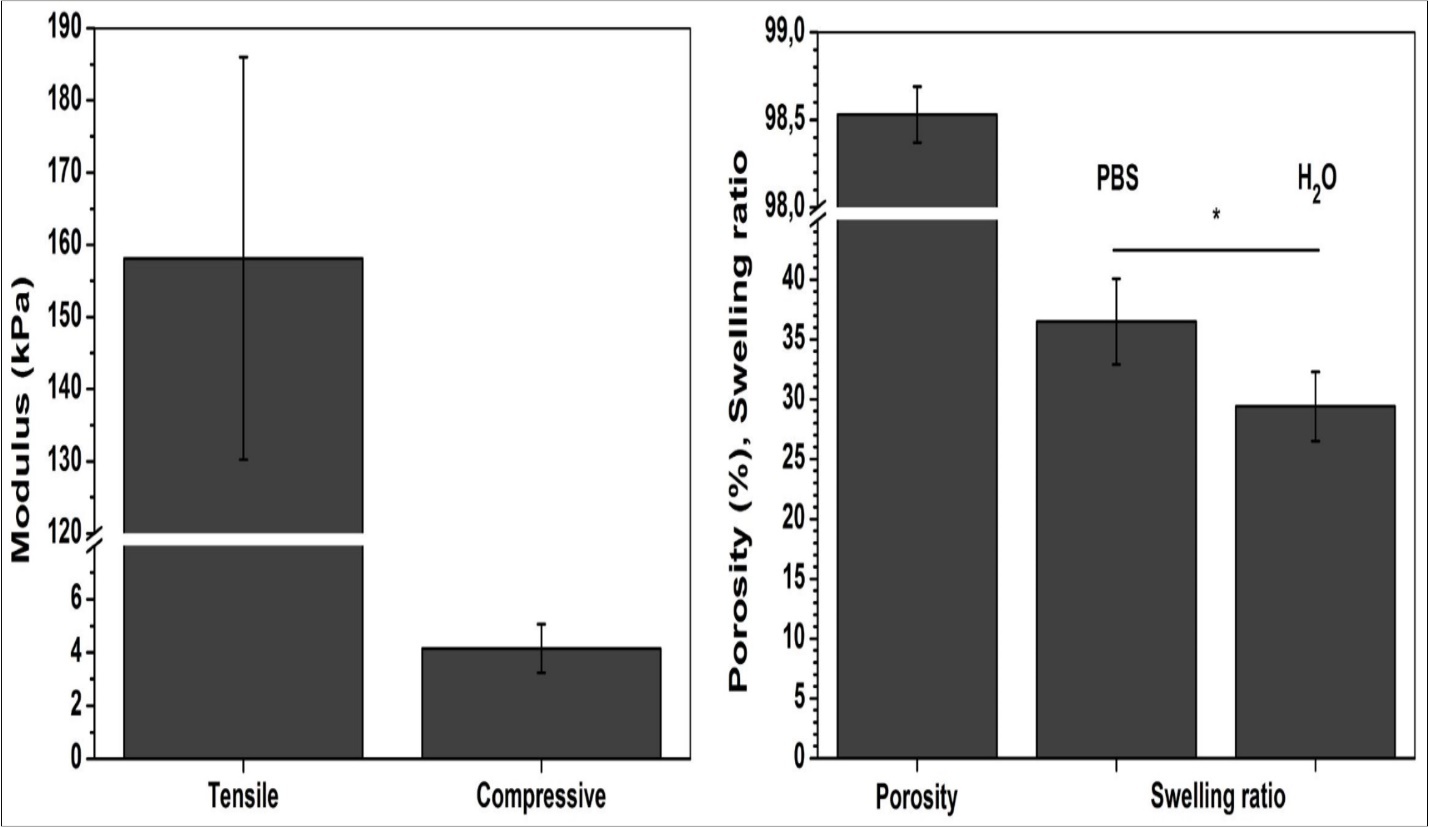
Chitosan has been extensively characterized by compressive and tensile test 19, 20, indicating its greater resistance when subjected to traction. In this study, previously swollen chitosan scaffolds were tested in wet state allowing elastic behaviour of chitosan. During swelling, chitosan spongy-like texture transforms to hydrocolloid with greater ability to deform. This is clearly visible from 40-fold difference of elastic modulus regarding to the one determined under compression.
Degradation Behaviour of the Scaffold
The effect of enzymatic degradation on microstructure of chitosan scaffolds was analysed by SEM. Morphology of the surface and cross section of chitosan scaffolds incubated four weeks in PBS and water containing lysozyme is given in Figure 3. According to the micrographs, significant difference in the material treated in LZ/PBS and LZ/H2O was not observed. However, CHT scaffold treated in LZ/PBS exhibit more irregular pore boundaries with signs of breakdown in the continuity of honey-comb like microstructure, with respect to the scaffold incubated in LZ/H2O. Moreover, sheet-like structure appeared on the surface of scaffold after 21 days in LZ/PBS, which has been previously reported by Qasim et al.21. After four weeks, the formation of large pores was evident on scaffolds incubated in both degradation media with greater pore roughness and discontinuity with respect to the initial scaffold.
Figure 3.Microstructure of chitosan cross section (left) and surface (right) after four weeks of incubation in different lysozyme solutions.
To support observations provided by electron microscopy, we investigated the degradation behaviour using gravimetric method. According to the literature1, the weight loss of porous chitosan depends on the initial concentration of chitosan solution, degree of deacetylation, distribution of molecular weight and swelling properties. Since we investigated chitosan materials with very high deacetylation degree (95 – 98%), it is no surprise to obtain lower weight loss due to the low sensitivity of amine groups to lysozyme. The degradation degree (Δm) of chitosan scaffolds incubated in LZ/PBS and LZ/H2O is depicted in Figure 4. The weight loss of chitosan scaffold (Figure 4a) exhibit the highest value of 17.3 ± 0.8 % after four weeks of incubation in LZ/PBS. On the other hand, weight loss resulted from the LZ/H2O incubation (Figure 4b), shows maximum value of 13.0 ± 1.9 % after first week, which is higher than scaffold incubated in LZ/PBS at the same time point (8.3 ± 1.2 %). The control samples incubated in PBS and H2O without lysozyme showed a similar trend of losing weight with approximately seven percent of total loss after first week, which remains constant during degradation study.
Figure 4.Degradation ratio of chitosan scaffolds performed in phosphate buffer saline solution a) and water b); the pH of incubation medium during degradation c).
To gather more insight into degradation behaviour, we monitored the pH of incubation media (Figure 4c). The buffered-medium (LZ/PBS and PBS) did not show any changes during total incubation period; however, there are certain oscillations in water with and without lysozyme (LZ/H2O and H2O). Initial pH value of the aqueous solution with lysozyme is 3.7, which is lower than pK of chitosan (6.5). After first week of degradation, initial pH of LZ/H2O has increased and maintained over pH of 4. This could be a consequence of chitosan dissolution behaviour. Being polycation, chitosan is soluble in acid environment where functional amine (–NH2) groups neutralise the hydrogen ion (–NH3+) originated from the dissociation of the acid. In this way, chitosan acts as base increasing the pH value. Significant increase in pH of LZ/H2O during four weeks of incubation with respect to the initial solution can indicate that dissolution of chitosan scaffold was maintained by bi-weekly refreshment of lysozyme solution. The alternate change in pH of LZ/H2O supernatant could be a result of gradual dissolution of chitosan which is indicated by the weight loss. Such behaviour can originate from broad distribution of molecular weight (100 000 – 300 000) and of pore size (100 – 250 µm). In contrast to the LZ/H2O medium, water control medium exhibited higher pH with no significant difference in weight loss after first week indicating lower chitosan dissolution.
The strange degradation behaviour of chitosan scaffolds in lysozyme containing water medium has led us to identify the organic compounds of degraded-chitosan samples. Figure 5 represents FTIR spectra of initial chitosan scaffolds, lysozyme and scaffolds degraded in LZ/PBS and LZ/H2O after 4 weeks of incubation.
Figure 5.FTIR spectra of initial chitosan scaffold (CHT), lysozyme powder (LZ) and chitosan scaffolds during four weeks of incubation in phosphate buffer saline solution (PBS) and water with or without lysozyme.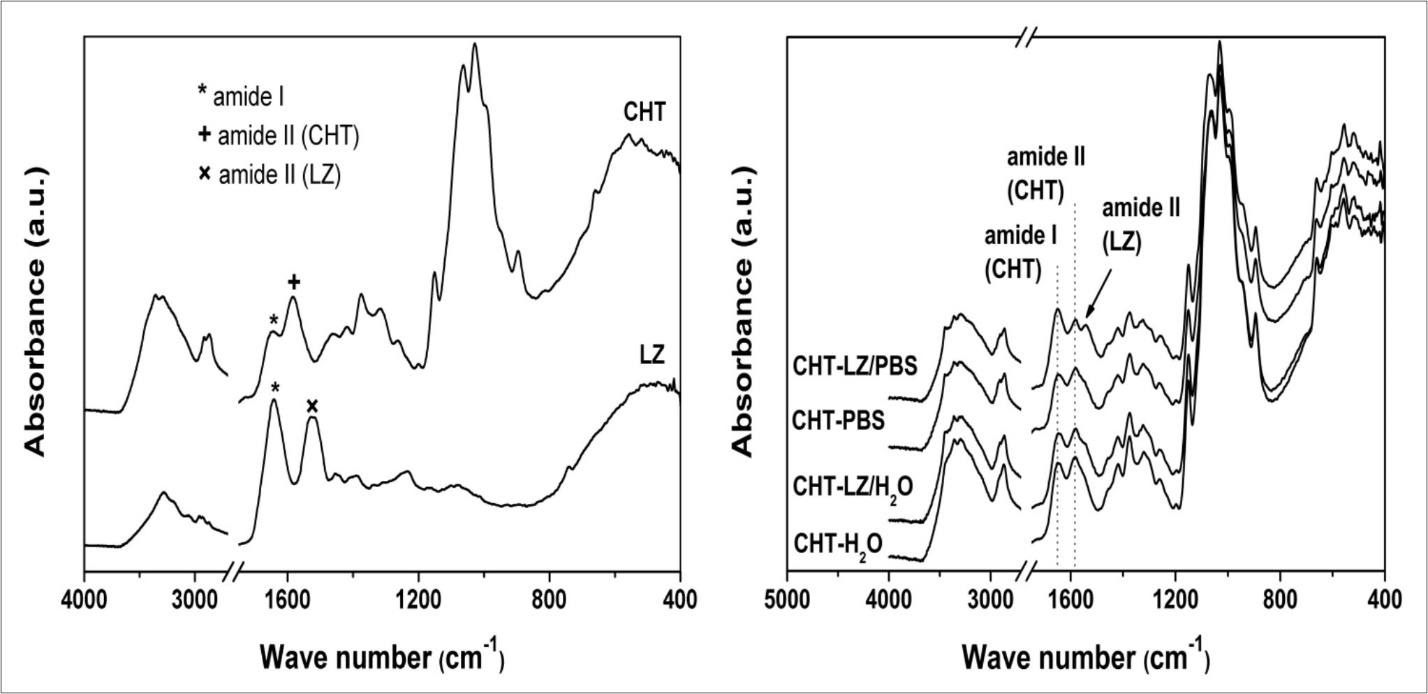
The characteristic absorption bands of chitosan corresponds to the stretching of the carbonyl group (amide I) and −NH bending (amide II) at 1644 cm-1 and 1583 cm-1, respectively, whose intensity depends on the deacetylation degree. The sharpness of the band at 1583 cm-1 confirms high deacetylation degree of chitosan. The absorption band in the range of 3359 − 3290 cm-1 is assigned to the overlapping of stretching vibration of amine (–NH2) and hydroxyl (–OH) groups. The absorption bands at 2917 cm-1 and 2869 cm-1 are associated with symmetric and asymmetric stretching of –CH, –CH2OH and –CH3 groups of the saccharide ring. The bands at 1420 cm-1 and 1374 cm-1 belong to the C−N and –CH3 bond of the acetyl group. Three stretching –CO groups are founded at range of wave numbers of 1151 – 1028 cm-1: the band at 1151 cm-1 is attributed to the asymmetric stretching of the oxygen atom from the bridge and carbon atom from the ring, while the bands from 1063 cm-1 to 1028 cm-1 are associated with –COH, –COC and –CH2OH22, 23. The FTIR spectrum of lysozyme exhibits characteristic amide bands at 1640 cm-1 (C=O; amide I) and 1525 cm-1 (–NH2; amide II) of polypeptide chain24.
The FTIR analysis of chitosan scaffolds degraded in LZ/PBS medium (CHT-LZ/PBS) showed the presence of amide absorption band of lysozyme which can indicate its remained deposition, compared to the control samples incubated in PBS. Even though the degraded samples were extensively washed with distilled water, the characteristic absorption band of lysozyme confirms strong electrostatic interactions between chitosan and lysozyme, which are necessary for glycoside bond cleavage. Interestingly, scaffolds that were subjected to the enzymatic degradation in water (CHT-LZ/H2O) show no difference in the FTIR spectrum with respect to the control scaffolds (CHT- H2O) indicating the lack of lysozyme activity in the pH range of 4 – 5, and chitosan dissolution process as a mechanism of scaffold’s weight loss.
Discussion
Chitosan is one of widely studied implantable materials for regenerative medicine and tissue engineering, mostly for wound and bone tissue restoration25, 26, 27. Among important requirements (biocompatibility, non-immunogenic properties, high porosity, and resistance to different mechanical forces) artificial graft has to be biodegradable with degradation rate that follows the rate of new tissue formation28. The degradation process is dictated by the mechanism of molecule cleavage into smaller ones and finally into molecules degradable by the biological processes. Accordingly, the degradation rate depends on materials properties such as the nature and composition, crystallinity and molecular weight, porosity, the pH of environment and implantation site. The insight into materials behaviour under physiological conditions will guide the design and development of potential tissue substituent or drug delivery system in terms of porosity, mechanical stability, surface modification, the release rate of active substance etc.
In this study, we investigated the degradation behaviour of chitosan scaffolds commonly used in tissue engineering. The main objective was to elucidate how medium conditions, supported by lysozyme presence, influence the chitosan biodegradation. Since prepared chitosan scaffolds are highly porous with good pore interconnectivity, the enzyme transport and diffusion of degradation products from material is facilitated, which is essential for tissue restoration. During enzymatic degradation, an enzyme first diffuses from the surrounding solution to the surface of the material. The subsequent adsorption leads to the formation of the enzyme-material complex. Then, enzyme starts the catalysis reaction of macromolecule cleavage causing the release of degradation products into the solution29, 30. A two-stage degradation of chitosan scaffolds with greater weight loss was observed in LZ/PBS medium. Low degradation degree of prepared chitosan is associated with very low quantity of acetyl group which are responsible for lysozomal binding20. The weight loss of chitosan scaffolds is generated by both, the lysozyme activity and chitosan dissolution. It is well known that chitosan dissolves when pH is lower than 6.5. However, the dissolution properties depend on molecular weight and MW distribution which is wide for chitosan used in this study. Therefore, chitosan dissolution could be possible even at neutral pH.
On contrary, the absence of significant difference in chitosan weight loss obtained from the LZ/H2O and H2O incubation can be related to the lysozyme inactivity. According to the manufacturer’s data sheet, lysozyme activity depends on the pH of dissolving medium, i.e. it is active at pH from 6.0 to 9.0. As shown from pH measurements, initial pH of LZ/H2O solution was far from the lysozyme activation conditions which solely resulted in dissolution process. This assumption was also confirmed by FTIR identification. As aforementioned, specific interactions between chitosan and lysozyme have to be provided to cleave the glycoside bond of chitosan. The absence of adsorbed lysozyme on scaffolds incubated in LZ/H2O medium confirms its inactivity. The chitosan weight loss in water containing lysozyme was not provided by the chemical reactions occurring during in vitro degradation. However, Han et al.13 studied the lysozyme degradation in acetate buffer (pH = 4.5) obtaining higher degradation and reduction of sugar ends regarding the phosphate buffered lysozyme solution and other buffered solution with pH of 4.5. Those results are in conflict with the fact that chitosan forms a chitosan-acetate buffer solution at pH of 4.5 – 4.8. Unfortunately, the mentioned study lacks information of lysozyme activity and identification of organic components of degraded samples.
The versatile application of chitosan-based materials in biomedical and pharmaceutical sciences imposes careful choosing of chitosan molecular weight, deacetylation degree and crystallinity that dictates in vitro and in vivo degradation. The identification of in vitro biodegraded chitosan materials represents great importance for designing and development of artificial grafts and drug delivery systems. Additionally, special attention should be given to degradation parameters such as pH of the medium and behaviour of applied enzyme. Moreover, we showed that most commonly used treatment of washing after scaffolds degradation is not adequate for determination of weight loss by gravimetric method due to strong interactions of lyosozyme active sites and N-acetlyglucosamine units of chitosan.
Degradation behaviour of potential tissue substituent or drug delivery system plays a role in treating the tumour-tissues. The pH of solid tumours is acidic due to increased fermentative metabolism which makes the adjacent normal tissue also acidic, leading to tumour invasion31. Chitosan-based nanoparticles showed their potential as drug-delivery carriers for cancer therapy due to the improvement of pharmacological and therapeutic properties of anti-cancer drugs controlling their release rate32. Since the peritumoral pH is heterogeneous, its direct monitoring near the treated surface in vivo could elucidate the microenvironmental conditions by pH microelectrodes33. The findings brought up by this study could help to determine if the rate of drug release and implant weight loss in similar applications is governed by the chitosan dissolution or biodegradation process.
Conclusions
The chitosan-based materials show promising potential in biomedical and pharmaceutical applications which requires suitable characterisation depending on the specific application. The degradation studies of chitosan scaffolds indicated limited activity of lysozyme which depends on the pH of incubation medium. According to the gravimetric analysis and FTIR identification, in vitro enzymatic degradation performed in phosphate buffer saline solution is dictated by physical (dissolution) and chemical (cleavage of glycoside bonds) processes, while lysozyme containing water medium causes only dissolution of chitosan. So far, such confirmation of enzyme inactivity has not yet been observed.
Acknowledgments
The Croatian Science Foundation under the project IP-2014-09-3752 is gratefully acknowledged. The authors want to thank Prof. G. Gallego Ferrer from Centre of Biomaterials and Tissue Engineering, Polytechnic University of Valencia, Spain for helping with compressive and tensile test.
References
- 3.Wiśniewska-Wrona M, Niekraszewicz A, Ciechańska D, Pospieszny H, Orlikowski L B. (2007) . , Polish Chitin Society 12, 149-156.
- 5.Wedmore I, J G McManus, Pusateri A E, Holcomb J B. (2006) . , The Journal of Trauma, Injury, Infection, and Critical Care 3, 655-658.
- 10.Li Q, Dunn E T, Grandmaison E W, Goosen MFA. (1992) . , Journal of Bioactive and Compatible Polymers 7, 370-397.
- 11.Beppu M, Vieira M, R S, Aimoli C G, Santana C C. (2007) . , Journal of Membrane Science 301, 126-130.
- 13.Han T, Nwe N, Furuike T, Tokura S, Tamura H. (2012) . , Journal Biomedical Science and Engineering 5, 15-23.
- 18.Rogina A, Rico P, Ferrer Gallego, Ivanković G, Ivanković M et al. (2015) . , European Polymer Journal 68, 278-287.
- 19.Albanna M Z, Bou-Akl T H, IIIHL Walters, Matthew HWT. (2012) . , Journal of Mechanical Behavior of Biomedical Materials 5, 171-180.
- 21.Qasim S B, Husain S, Huang Y, Pogorielov M, Deineka V et al. (2017) . Polymer Degradation and Stability 136 31-38.
- 22.Duarte M L, Ferreira M C, Marvão M R, Rocha J. (2002) . , International Journal of Biological Macromolecules 31, 1-8.
- 24.Sowjanya J, Singh A, Mohita J, Sarvanan T, Moorthi S et al. (2013) . Colloids and Surfaces B: Biointerfaces 109 294-300.
- 26.Jiang T, Nukavarapu S P, Deng M, Jabbarzadeh E, Kofron. (2010) . , Acta Biomaterialia 6, 3457-3470.
- 30.Azevedo H S, Reis R L. (2005) Understanding the enzymatic degradation of biodegradable polymers and strategies to control their degradation rate, in:. Biodegradable Systems in Tissue Engineering and Regenerative Medicine, CRC Press 12 , USA .
Cited by (248)
This article has been cited by 248 scholarly works according to:
Citing Articles:
Carbohydrate Polymers (2025) Crossref
Carbohydrate Polymers (2025) OpenAlex
Vera-Maria Platon, Bianca-Iustina Andreica, Alexandru Anisiei, I. Roșca, I. Sandu et al. - Carbohydrate Polymers (2025) Semantic Scholar
Heru Agung Saputra, Andreas - Next Materials (2025) Semantic Scholar
International Journal of Biological Macromolecules (2025) OpenAlex
International Journal of Biological Macromolecules (2025) Crossref
Olunusi Samuel Olugbenga, Ramli Nor Hanuni, Adam Fatmawati, Emran Hossain, Tak H Kim - International Journal of Biological Macromolecules (2025) Semantic Scholar
International Journal of Biological Macromolecules (2025) Crossref
International Journal of Biological Macromolecules (2025) OpenAlex
Fatmagul Gedik, Turdimuhammad Abdullah, Şerife Tozan Rüzgar, Funda Keteci, Buket Bakan et al. - International Journal of Biological Macromolecules (2025) Semantic Scholar
T. Nguyen, Jun Seop Lee - Polymers (2025) Semantic Scholar
Alexandra I. F. Alves, N. Alves, Juliana R. Dias - Polymers (2025) Semantic Scholar
Kamini Sahu, Anjila Firdous, Mohammad Adnan Raza, Suprit D. Saoji, Vandana B. Patravale et al. - Journal of Drug Delivery Science and Technology (2025) Semantic Scholar
Journal of Drug Delivery Science and Technology (2025) OpenAlex
Journal of Drug Delivery Science and Technology (2025) OpenAlex
Ahmed M. Omer, Eman M. Abd El-Monaem, Heba S. Hassan, Mahmoud A. Elmeligy, Angela Kleinová et al. - Journal of Drug Delivery Science and Technology (2025) Semantic Scholar
International Journal of Biological Macromolecules (2025) OpenAlex
M. Iftime, G. Ailiesei, Simona Morariu, Andreea-Isabela Sandu, Cristina Mihaela Râmbu et al. - International Journal of Biological Macromolecules (2025) Semantic Scholar
Varshan Gounden, Moganavelli Singh - Pharmaceutics (2025) Semantic Scholar
International Journal of Biological Macromolecules (2025) OpenAlex
Sandu Cibotaru, Alexandru Anisiei, Vera-Maria Platon, I. Roșca, I. Sandu et al. - International Journal of Biological Macromolecules (2025) Semantic Scholar
RSC Advances (2025) OpenAlex
RSC Advances (2025) Crossref
Argelia Rosillo-de la Torre, Tonantzi Pérez-Moreno, I. Quintero-Ortega, P. Sahare, D. Bravo-Alfaro et al. - RSC Advances (2025) Semantic Scholar
Advanced Materials Technologies (2025) Crossref
Polymer (2025) OpenAlex
Tanawat Buntum, Nanthanit Jaruseranee, Mattaka Kongkow, U. Ruktanonchai, Hasan Uludağ et al. - Polymer (2025) Semantic Scholar
Journal of Porous Materials (2025) Crossref
Maria Diva Rossary, Jeniffer Kalyana Paramita Djunaedi, Teresa Prinasti Riyantoro, Agustina Setiawati - TRENDS IN THE SCIENCES (2025) Semantic Scholar
Drug Delivery and Translational Research (2025) OpenAlex
Drug Delivery and Translational Research (2025) Crossref
André Miguel Martinez Junior, T. F. Ruiz, P. S. Vilamaior, V. Tiera, S. Taboga et al. - Drug Delivery and Translational Research (2025) Semantic Scholar
Engineered Regeneration (2024) Crossref
Engineered Regeneration (2024) OpenAlex
Yang Yu, Yinxiang Tang, Weiwen Liang, Yuanbin Wang, Ouyang Yang et al. - Engineered Regeneration (2024) Semantic Scholar
Milena Costa da Silva Barbosa, H. Silva, Débora de Sousa Lopes, W. F. Wanderley, Rosana Araújo Rosendo et al. - Materials Research (2024) Semantic Scholar
Materials Research (2024) OpenAlex
ACS Biomaterials Science & Engineering (2024) OpenAlex
ACS Biomaterials Science & Engineering (2024) Crossref
Siena M. Mantooth, Asher M Hancock, Peter M Thompson, George Varghese P J, Danielle M. Meritet et al. - ACS Biomaterials Science & Engineering (2024) Semantic Scholar
Alyeh Abdollahi, H. Aghayan, Zahra Mousivand, Hamidreza Motasadizadeh, Samane Maghsoudian et al. - Stem cell research & therapeutics (2024) Semantic Scholar
Journal of Composites Science (2024) OpenAlex
Lya Piaia, S. Silva, E. M. Fernandes, J. M. Gomes, A. Franco et al. - Journal of Composites Science (2024) Semantic Scholar
Polymer Engineering and Science (2024) OpenAlex
Leila Soltani, Kambiz Varmira, Maryam Nazari - Scientific Reports (2024) Semantic Scholar
Journal of Drug Delivery Science and Technology (2024) Crossref
Journal of Drug Delivery Science and Technology (2024) OpenAlex
Syed Ali Faran, Dan Chau Thuy Nguyen, Joseph Dowling, Richie Ryan, Peter Mcloughlin et al. - Journal of Drug Delivery Science and Technology (2024) Semantic Scholar
Journal of Biomaterials Science, Polymer Edition (2024) Crossref
Journal of Biomaterials Science Polymer Edition (2024) OpenAlex
G. Navidi, S. Same, M. Allahvirdinesbat, Parvaneh Nakhostin Panahi, Kazem Dindar Safa - Journal of Biomaterials Science. Polymer Edition (2024) Semantic Scholar
Journal of Applied Polymer Science (2024) OpenAlex
Journal of Applied Polymer Science (2024) Crossref
Gizem Baysan, Pınar Akokay Yılmaz, R. Husemoglu, A. Z. Albayrak, A. Şişman et al. - Journal of Applied Polymer Science (2024) Semantic Scholar
Polymers (2024) OpenAlex
Yulia V Zhuikova, V. Zhuikov, D. Khaydapova, A. P. Lunkov, G. Bonartseva et al. - Polymers (2024) Semantic Scholar
Nanomedicine (2024) Crossref
Nanomedicine (2024) OpenAlex
Jia Sheng Yew, S. Ong, Hui Xuan Lim, Soon Hao Tan, K. Ong et al. - Nanomedicine (2024) Semantic Scholar
Advanced Materials Technologies (2024) OpenAlex
R. N. F. Moreira Filho, Matheus Xavier de Oliveira, Ana Lorena Brito Soares, Lidyane Souto Maciel Marques, Pascale Chevallier et al. - Advanced Materials & Technologies (2024) Semantic Scholar
Polymers (2024) OpenAlex
Oana-Teodora Afloarea, Isabella Nacu, L. Vereștiuc, C. Yilmaz, A. Panainte et al. - Polymers (2024) Semantic Scholar
Biomedical Materials (2024) Crossref
Biomedical Materials (2024) OpenAlex
Katherine A Pitrolino, R. Felfel, George Roberts, C. Scotchford, David M. Grant et al. - Biomedical Materials (2024) Semantic Scholar
Chemistry of Materials (2024) OpenAlex
Chemistry of Materials (2024) Crossref
María Luisa Del Pozo, Antonio Aguanell, E. García-Junceda, Julia Revuelta - Chemistry of Materials (2024) Semantic Scholar
Frontiers in Bioengineering and Biotechnology (2024) OpenAlex
Frontiers in Bioengineering and Biotechnology (2024) Crossref
Aref Yarahmadi, Behrooz Dousti, Mahdi Karami-Khorramabadi, Hamed Afkhami - Frontiers in Bioengineering and Biotechnology (2024) Semantic Scholar
Polymers (2024) OpenAlex
Juan Hernández, Concepción Panadero-Medianero, Macarena S. Arrázola, Manuel Ahumada - Polymers (2024) Semantic Scholar
Journal of the mechanical behavior of biomedical materials/Journal of mechanical behavior of biomedical materials (2024) OpenAlex
M. Jaramillo, Cristian Covarrubias, Edwin Patiño González, C. O. Ossa Orozco - Journal of The Mechanical Behavior of Biomedical Materials (2024) Semantic Scholar
International Journal of Biological Macromolecules (2024) OpenAlex
Ahmed M. Omer, M. A. Elmeligy, Eman M. Abd El-Monaem, Basma H. Naiel, M. Barlog et al. - International Journal of Biological Macromolecules (2024) Semantic Scholar
C. S. Cabral, Duarte de Melo-Diogo, Paula Ferreira, A. Moreira, I. Correia - International Journal of Biological Macromolecules (2024) Semantic Scholar
International Journal of Biological Macromolecules (2024) OpenAlex
International Journal of Biological Macromolecules (2024) Crossref
Macromolecular Chemistry and Physics (2024) Crossref
Macromolecular Chemistry and Physics (2024) OpenAlex
Nano Energy (2024) OpenAlex
G. de Marzo, V. Mastronardi, M. Todaro, Laura Blasi, Valentina Antonaci et al. - Nano Energy (2024) Semantic Scholar
Journal of Porous Materials (2024) OpenAlex
L. R. M. de Andrade, W. S. Mota, Raquel de Melo Barbosa, J. Cardoso, Luciana N. Andrade et al. - Journal of porous materials (2024) Semantic Scholar
International Journal of Biological Macromolecules (2024) OpenAlex
Sadaf Nosheen, Hamid Mukhtar, Sajjad Haider, Rawaiz Khan, Faiza Sharif - International Journal of Biological Macromolecules (2024) Semantic Scholar
Biomedical Materials (2024) OpenAlex
Biomedical Materials (2024) Crossref
Tariq O. Abbas, Hemalatha Parangusan, Huseyin C. Yalcin, Mohammad K. Hassan, Lubna Zakrif et al. - Biomedical Materials (2024) Semantic Scholar
ACS Sustainable Resource Management (2024) Crossref
ACS Sustainable Resource Management (2024) OpenAlex
Sofie E. Svensson, Mehdi Abdollahi, F. Moghadam, N. Kalita, M. Hakkarainen et al. - ACS Sustainable Resource Management (2024) Semantic Scholar
Saeeun Jang, Sukho Park - Sensors and Actuators B: Chemical (2023) Semantic Scholar
Sensors and Actuators B Chemical (2023) OpenAlex
International Journal of Biological Macromolecules (2023) OpenAlex
Xuehua Li, Yali Liu, Hongrong Song, Meiting Zhao, Qin Song - International Journal of Biological Macromolecules (2023) Semantic Scholar
Liaw Hui Zoe, S. David, Rajan Rajabalaya - Toxicology Reports (2023) Semantic Scholar
Toxicology Reports (2023) OpenAlex
A. Pádua, L. Figueiredo, J. Silva, J. Borges - Progress in Biomaterials (2023) Semantic Scholar
Progress in Biomaterials (2023) OpenAlex
Progress in Biomaterials (2023) Crossref
A. Ressler, Roope Ohlsbom, Andreja Žužić, Arjen Gebraad, Erkka J. Frankberg et al. - European Polymer Journal (2023) Semantic Scholar
European Polymer Journal (2023) OpenAlex
Luca Signorini, G. Marenzi, Anastasia Facente, B. Marrelli, Rosa Maria Marano et al. - International Journal of Medical Sciences (2023) Semantic Scholar
International Journal of Medical Sciences (2023) OpenAlex
Advanced Healthcare Materials (2023) OpenAlex
Advanced Healthcare Materials (2023) Crossref
International Journal of Pharmaceutics (2023) OpenAlex
Delia Mihaela Raţă, Anca Niculina Cadinoiu, Leonard Ionut Atanase, Gabriela Calin (Mihalache), M. Popa - International journal of pharmaceutics (2023) Semantic Scholar
T. Bharadwaj, Shreya Chrungoo, D. Verma - Journal of Biomaterials Science. Polymer Edition (2023) Semantic Scholar
Journal of Biomaterials Science Polymer Edition (2023) OpenAlex
Journal of Biomaterials Science, Polymer Edition (2023) Crossref
Industrial Crops and Products (2023) OpenAlex
Kartikey Verma, S. Siddiki, C. Maity, Raghvendra Kumar Mishra, M. Moniruzzaman - Industrial crops and products (Print) (2023) Semantic Scholar
Ali Deniz Dalgic, D. Atila, A. Tezcaner, S. Gürses, D. Keskin - European Polymer Journal (2023) Semantic Scholar
European Polymer Journal (2023) OpenAlex
European Polymer Journal (2023) Crossref
Sophie L. Reay, E. L. Jackson, Daniel Salthouse, A. Ferreira, C. Hilkens et al. - Polymers (2023) Semantic Scholar
Polymers (2023) OpenAlex
Crystals (2023) OpenAlex
M.I. Ayub, Mahwish Bashir, F. Majid, Rabia Shahid, Babar Shahzad Khan et al. - Crystals (2023) Semantic Scholar
Molecular Pharmaceutics (2023) Crossref
Molecular Pharmaceutics (2023) OpenAlex
Raquel de Castro, Gurshagan Kandhola, Jin-Woo Kim, Quincy C Moore, Audie K Thompson - Molecular Pharmaceutics (2023) Semantic Scholar
(2023) OpenAlex
G. Dalei, Subhraseema Das, Soumya Ranjan Jena, D. Jena, Jasmine Nayak et al. - Chemical Engineering and Science (2023) Semantic Scholar
Chemical Engineering Science (2023) OpenAlex
Priyanka Agarwal, I. Rupenthal - Advanced Drug Delivery Reviews (2023) Semantic Scholar
Advanced Drug Delivery Reviews (2023) OpenAlex
Advanced Drug Delivery Reviews (2023) Crossref
S. Ganesh, R. Anushikaa, Venkadesan Sri, Swetha Victoria, Krishnaraj Lavanya et al. - Journal of Functional Biomaterials (2023) Semantic Scholar
Journal of Functional Biomaterials (2023) OpenAlex
International Journal of Biological Macromolecules (2023) OpenAlex
S. Lekhavadhani, A. Shanmugavadivu, N. Selvamurugan - International Journal of Biological Macromolecules (2023) Semantic Scholar
E. Dresvyanina, N. A. Tagandurdyyeva, V. Kodolova-Chukhontseva, I. P. Dobrovol'skaya, A. Kamalov et al. - Polymers (2023) Semantic Scholar
Polymers (2023) OpenAlex
Inês Guimarães, Raquel Costa, Sara Madureira, S. Borges, A. Oliveira et al. - International Journal of Molecular Sciences (2023) Semantic Scholar
International Journal of Molecular Sciences (2023) OpenAlex
S. Patra, Monika Singh, Divya Pareek, Kirti Wasnik, Premshankar Gupta et al. - Reference Module in Materials Science and Materials Engineering (2022) Semantic Scholar
(2022) Crossref
Elsevier eBooks (2022) OpenAlex
G. Acik, C. Altinkok, B. Acik - Polymer Bulletin (2022) Semantic Scholar
Carbohydrate Polymers (2022) OpenAlex
Carbohydrate Polymers (2022) Crossref
Antonio Aguanell, María Luisa Del Pozo, Carlos Pérez-Martín, Gabriela Pontes, Á. Bastida et al. - Carbohydrate Polymers (2022) Semantic Scholar
C. Cannavà, F. De Gaetano, R. Stancanelli, V. Venuti, G. Paladini et al. - Pharmaceutics (2022) Semantic Scholar
Pharmaceutics (2022) OpenAlex
Abul K. Mallik, Adib H. Chisty, Sumaya F. Kabir, M. N. Khan, Papia Haque et al. - Chitosan in Biomedical Applications (2022) Semantic Scholar
International Journal of Biological Macromolecules (2022) OpenAlex
Md Sowaib Ibne Mahbub, S. Bae, J. Gwon, Byong-Taek Lee - International Journal of Biological Macromolecules (2022) Semantic Scholar
Prutha Joshi, Md Shakir Uddin Ahmed, K. Vig, M. Auad - Sustainable Chemistry (2022) Semantic Scholar
Sustainable Chemistry (2022) OpenAlex
A. Petkova, I. Kubajewska, Alexandra Vaideanu, A. Schätzlein, I. Uchegbu - Pharmaceutics (2022) Semantic Scholar
Pharmaceutics (2022) OpenAlex
Bioengineering & Translational Medicine (2022) Crossref
Carbohydrate Polymers (2022) OpenAlex
Carbohydrate Polymers (2022) Crossref
B. N. Kumara, R. Shambhu, A. Prabhu, K. S. Prasad - Carbohydrate Polymers (2022) Semantic Scholar
Macromolecular Bioscience (2022) Crossref
Macromolecular Bioscience (2022) OpenAlex
International Journal of Biological Macromolecules (2022) OpenAlex
Sheila Maiz-Fernández, L. Pérez-Álvarez, U. Silvan, J. Vilas‐Vilela, S. Lanceros‐Méndez - International Journal of Biological Macromolecules (2022) Semantic Scholar
Brian C. W. Webb, S. Rafferty, A. Vreugdenhil - Polymers (2022) Semantic Scholar
Polymers (2022) OpenAlex
International Journal of Biological Macromolecules (2022) OpenAlex
A. George, P. Shrivastav - International Journal of Biological Macromolecules (2022) Semantic Scholar
Macromolecular Chemistry and Physics (2022) Crossref
Macromolecular Chemistry and Physics (2022) OpenAlex
Carbohydrate Polymers (2022) OpenAlex
Carbohydrate Polymers (2022) Crossref
Khaleel A. Abu-sbeih, G. Al-Mazaideh, W. Al-Zereini - Carbohydrate Polymers (2022) Semantic Scholar
Jin Sil Lee, Hyeryeon Oh, Sunghyun Kim, Jeung-Hoon Lee, Y. Shin et al. - Pharmaceutics (2021) Semantic Scholar
Pharmaceutics (2021) OpenAlex
Dania Adila Ahmad Ruzaidi, M. Mahat, Saiful Arifin Shafiee, Zarif Mohamed Sofian, Awis Sukarni Mohmad Sabere et al. - Polymers (2021) Semantic Scholar
Polymers (2021) OpenAlex
Macromolecular Bioscience (2021) Crossref
Carbohydrate Polymers (2021) OpenAlex
Carbohydrate Polymers (2021) Crossref
Nishat Tabassum, Shoeb Ahmed, M. Ali - Carbohydrate Polymers (2021) Semantic Scholar
International Journal of Biological Macromolecules (2021) OpenAlex
N. Dabholkar, Srividya Gorantla, Sudhanshu Sharma, V. Rapalli, A. Alexander et al. - International Journal of Biological Macromolecules (2021) Semantic Scholar
Biomedical Materials (2021) Crossref
International Journal of Biological Macromolecules (2021) OpenAlex
R. Lungu, Alexandru Anisiei, I. Roșca, Andreea-Isabela Sandu, D. Ailincăi et al. - Reactive & functional polymers (2021) Semantic Scholar
Reactive and Functional Polymers (2021) OpenAlex
Journal of the mechanical behavior of biomedical materials/Journal of mechanical behavior of biomedical materials (2021) OpenAlex
Journal of the Mechanical Behavior of Biomedical Materials (2021) Crossref
Sabia Kouser, Sareen Sheik, A. Prabhu, G. K. Nagaraja, Kalappa Prashantha et al. - Journal of The Mechanical Behavior of Biomedical Materials (2021) Semantic Scholar
Jiyoo Baek, Mohankandhasamy Ramasamy, Natasha Carly Willis, D. Kim, W. Anderson et al. - Current Research in Food Science (2021) Semantic Scholar
Current Research in Food Science (2021) OpenAlex
Current Research in Food Science (2021) Crossref
Carbohydrate Polymers (2021) Crossref
Sita Shrestha, B. Shrestha, S. Ko, Rupesh Kandel, C. Park et al. - Carbohydrate Polymers (2021) Semantic Scholar
C. Andrițoiu, C. Andriescu, M. Danu, C. Lungu, B. Ivănescu et al. - Pharmaceuticals (2021) Semantic Scholar
Pharmaceuticals (2021) OpenAlex
N. Vo, Lei Huang, H. Lemos, A. Mellor, K. Novakovic - Journal of Applied Polymer Science (2021) Semantic Scholar
Journal of Applied Polymer Science (2021) OpenAlex
Journal of Applied Polymer Science (2021) Crossref
Vladyslav Vivcharenko, M. Wójcik, K. Pałka, Agata Przekora - Materials (2021) Semantic Scholar
Materials (2021) OpenAlex
Pedro L. Arruebo-Rivera, Freddy Castillo-Alfonso, Amanda Troya, Yosberto Cárdenas-Moreno, Patricia Pérez-Ramos et al. - The Protein Journal (2021) Semantic Scholar
The Protein Journal (2021) OpenAlex
RSC Advances (2021) OpenAlex
Debyashreeta Barik, K. Kundu, M. Dash - RSC Advances (2021) Semantic Scholar
Liming Zhou, Nanbo Liu, Longbao Feng, Mingyi Zhao, Peng Wu et al. - Bioengineering & Translational Medicine (2021) Semantic Scholar
Bioengineering & Translational Medicine (2021) OpenAlex
Carolina A. M. Ferreira, Adriana P. Januário, Rafael Félix, N. Alves, M. Lemos et al. - Pharmaceutics (2021) Semantic Scholar
Pharmaceutics (2021) OpenAlex
Jaundrie Fourie, Francois Taute, L. D. du Preez, D. D. de Beer - Journal of bioactive and compatible polymers (Print) (2021) Semantic Scholar
S. N. Jayash, P. Cooper, R. Shelton, S. Kuehne, G. Poologasundarampillai - International Journal of Molecular Sciences (2021) Semantic Scholar
International Journal of Molecular Sciences (2021) OpenAlex
W. El-Sayed, J. Alkabli, Akram Aloqbi, R. F. Elshaarawy - (2021) Semantic Scholar
European Polymer Journal (2021) OpenAlex
A. Omer, Maha S. Ahmed, G. El-Subruiti, R. Khalifa, Abdelazeem S. Eltaweil - Pharmaceutics (2021) Semantic Scholar
Pharmaceutics (2021) OpenAlex
Biomedical Materials (2021) Crossref
Prutha Joshi, Md Shakir Uddin Ahmed, K. Vig, I. B. Vega Erramuspe, M. Auad - Polymers for Advanced Technologies (2021) Semantic Scholar
Polymers for Advanced Technologies (2021) OpenAlex
Dania Adila Ahmad Ruzaidi, M. Mahat, Zarif Mohamed Sofian, Nikman Adli Nor Hashim, Hazwanee Osman et al. - Polymers (2021) Semantic Scholar
Polymers (2021) OpenAlex
Maria Dennise Medeiros Macêdo, Breno de Medeiros Lucena, G. R. C. Cerqueira, W. Sousa, Thiago Cajú Pedrosa et al. - (2021) Semantic Scholar
Research Society and Development (2021) OpenAlex
Journal of Pharmaceutical Sciences (2020) OpenAlex
Julia C. Bulmahn, H. Kutscher, Katherine Cwiklinski, S. Schwartz, P. Prasad et al. - Journal of Pharmacy and Science (2020) Semantic Scholar
Liszt Y. C. Madruga, R. Balaban, K. Popat, M. Kipper - Macromolecular Bioscience (2020) Semantic Scholar
Macromolecular Bioscience (2020) OpenAlex
C. Palomino-Durand, Marco Lopez, P. Marchandise, B. Martel, N. Blanchemain et al. - Pharmaceutics (2020) Semantic Scholar
Pharmaceutics (2020) OpenAlex
Ya Li, Xiaonan Wang, Yuanyuan Han, Huanxiang Sun, J. Hilborn et al. - Biomedical Materials (2020) Semantic Scholar
Biomedical Materials (2020) OpenAlex
Qianyu Sun, Jie Sheng, Rendang Yang - (2020) Semantic Scholar
Polymer Degradation and Stability (2020) OpenAlex
Journal of Molecular Graphics and Modelling (2020) OpenAlex
Journal of Molecular Graphics and Modelling (2020) Crossref
D. Roman, V. Ostafe, A. Isvoran - Journal of Molecular Graphics and Modelling (2020) Semantic Scholar
International Journal of Biological Macromolecules (2020) OpenAlex
Vladyslav Vivcharenko, A. Benko, K. Pałka, M. Wójcik, Agata Przekora - International Journal of Biological Macromolecules (2020) Semantic Scholar
Muhammad Asim Akhtar, Z. Hadzhieva, I. Dlouhý, A. Boccaccini - Coatings (2020) Semantic Scholar
Coatings (2020) OpenAlex
Carbohydrate Polymers (2020) OpenAlex
G. Sharmila, C. Muthukumaran, S. Kirthika, Sundarapandian Keerthana, N. Kumar et al. - International Journal of Biological Macromolecules (2020) Semantic Scholar
International Journal of Biological Macromolecules (2020) OpenAlex
International Journal of Biological Macromolecules (2020) OpenAlex
International Journal of Biological Macromolecules (2020) Crossref
Sabia Kouser, Sareen Sheik, G. K. Nagaraja, A. Prabhu, Kalappa Prashantha et al. - International Journal of Biological Macromolecules (2020) Semantic Scholar
ACS Biomaterials Science & Engineering (2020) OpenAlex
ACS Biomaterials Science & Engineering (2020) Crossref
A. Ambrosone, L. Matteis, Inés Serrano-Sevilla, C. Tortiglione, J. M. de la Fuente - ACS Biomaterials Science & Engineering (2020) Semantic Scholar
Griselda V Nájera-Romero, M. Yar, I. Rehman - Functional Composite Materials (2020) Semantic Scholar
Functional Composite Materials (2020) Crossref
Functional Composite Materials (2020) OpenAlex
Shuxin Li, Jun Zhou, YiHui Huang, Joyita Roy, Ning Zhou et al. - Acta Biomaterialia (2020) Semantic Scholar
Acta Biomaterialia (2020) OpenAlex
Acta Biomaterialia (2020) Crossref
I. A. Kadhim, Z. A. Abdul Ameer, Aseel B. Alzubaidi - IOP Conference Series: Materials Science and Engineering (2020) Semantic Scholar
IOP Conference Series Materials Science and Engineering (2020) OpenAlex
Journal of Colloid and Interface Science (2020) OpenAlex
N. L. Benbow, Damien A. Sebben, S. Karpiniec, D. Stringer, M. Krasowska et al. - Journal of Colloid and Interface Science (2020) Semantic Scholar
Ewelina Jakubowska, M. Gierszewska, J. Nowaczyk, Ewa Olewnik-Kruszkowska - (2020) Semantic Scholar
Food Hydrocolloids (2020) OpenAlex
Satyavrat Tripathi, B. Singh, S. Divakar, G. Kumar, S. Mallick et al. - Biomedical Materials (2020) Semantic Scholar
Biomedical Materials (2020) OpenAlex
M. Bil, Izabela Hipś, P. Mrówka, W. Świȩszkowski - (2020) Semantic Scholar
Polymer Degradation and Stability (2020) OpenAlex
Carbohydrate Polymers (2020) OpenAlex
Carbohydrate Polymers (2020) Crossref
Sheila Maiz-Fernández, O. Guaresti, L. Pérez-Álvarez, L. Ruiz‐Rubio, N. Gabilondo et al. - Carbohydrate Polymers (2020) Semantic Scholar
Bernd C. Schmid - Der Gynäkologe (2019) Semantic Scholar
Der Gynäkologe (2019) Crossref
Der Gynäkologe (2019) OpenAlex
Ashraq A. Kadhm, Z. J. A. Alameer, A. A. Zubaidi - XIAMEN-CUSTIPEN WORKSHOP ON THE EQUATION OF STATE OF DENSE NEUTRON-RICH MATTER IN THE ERA OF GRAVITATIONAL WAVE ASTRONOMY (2019) Semantic Scholar
AIP conference proceedings (2019) OpenAlex
Carbohydrate Polymers (2018) OpenAlex
Carbohydrate Polymers (2018) Crossref
Karthik Rathinam, S. Singh, Christopher J. Arnusch, R. Kasher - Carbohydrate Polymers (2018) Semantic Scholar
V. Chander, A. K. Singh, G. Gangenahalli - Journal of Microencapsulation (2018) Semantic Scholar
Journal of Microencapsulation (2018) Crossref
Journal of Microencapsulation (2018) OpenAlex
G. Amariei, V. Kokol, K. Boltes, P. Letón, R. Rosal - RSC Advances (2018) Semantic Scholar
RSC Advances (2018) OpenAlex
RSC Advances (2018) Crossref
International Journal of Molecular Sciences (2018) OpenAlex
Cheng-Han Tsai, Peng-Yuan Wang, I-Chan Lin, Hu Huang, Guei-Sheung Liu et al. - International Journal of Molecular Sciences (2018) Semantic Scholar