
Molecular Spectroscopy and Molecular Docking Studies on (E)-1-(4-Bromobenzylidene) Thiourea
Abstract
The organic molecule (E)-1-(4-bromobenzylidene)thiourea (BBTU) have been synthesized and characterized using FT-IR and FT-Raman spectral studies. The quantum chemical calculations of BBTU have been studied using DFT/B3LYP/6-31G(d,p) level of theory. The stable conformer is identified by the potential energy surface scan. The complete vibrational assignments were performed on the basis of PED analysis with the help of SQM method. NBO analysis was carried out to explore the various conjucative/hyperconjucative interactions within the molecule and their second order stabilization energy. The NLO activity of BBTU is calculated and compared with the standard Urea molecule. The energies of the FMOs are used for the determination of global reactivity descriptors. The electrophilic and nucleophilic charge sites were identified by the molecular electrostatic potential mapped surface. The molecular docking of BBTU is carried out with the receptors of 3U2D and 1JIJ to screen the bacterial activity.
Article Information
- Received
- Accepted
- Published
Academic Editor: Praveen Kumar Sharma, Lovely Professional University, Phagwara, Punjab
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2018 Shyamal Das, et al.
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Corresponding author: S. Bharanidharan, Department of Physics, Bharath Institute of Higher Education and Research, Bharath University, Chennai-600 073, India —
Competing Interests
The authors have declared that no competing interests exist.
Funding
No specific funding statement was provided by the authors.
Data Availability
No data-availability statement was provided by the authors.
Citation:
Introduction
The consideration products of carbonyl compounds and primary amines are often named as Schiff bases. They are also known as azomethines or anils or imines. The >C=N-group is present in organic molecules of fundamental importance. They have got extensive application in biological and industrial fields. Schiff bases with potential pharmaceutical use were synthesized 1, 2. Schiff bases have been reported for their biological properties, such as anti-bacterial, anti-fungal, anti-inflammatory, analgesic, anti-convulsant, anti-tubercular, anti-cancer, anti-oxidant and anti-helmintic activities 3, 4, 5, 6, 7, 8, 9, 10, 11. Schiff base metal complexes have applications in the areas from material science to biological sciences. They have been widely studied because they have anti-cancer and herbicidal applications 12, 13. Schiff base complexes show greater biological activity than free ligands. The DNA binding, cytotoxicity and apoptosis induction activity were studied for Schiff base copper(II) complexes 14. A rather commonly used technique regards the development of fluorescent chemosensor quenching of fluorescence by interaction with anions. The sensors can provide fast and visible color changes from yellow to red in the presence of strong basic anions. Over the past 10 years, several excellent chemosensors have been reported for recognition and sensing of anions with high selectivity and sensitivity 15, 16, 17, 18, 19.
The uses of Schiff bases and their metal complexes as photovoltaic material has gained interest due to their easy synthesize procedure and complexation with metal ions. This quality of Schiff bases and their metal complexes make them as a potential substitute for organic solar cells. Research has been done with Schiff bases and their metal complexes as sensitizing and acceptor materials in dye sensitized solar cells DSSC, but their application in organic solar cells are not familiar. The high thermal stability, wider range of absorption, lower band gap and good electrical conductivity properties promises the use of Schiff bases and their metal complexes as the future materials for organic photovoltaics and the smaller band gap is considerable to improve the DSSC performance 20, 21.
Literature survey reveals that the propargyl arms containing Schiff base (L) was synthesized by the condensation of 1-2ethanone with trans-1,2-diaminocyclohexane by C. Balakrishnan et al (2015) 22. The single crystal analysis shows that the compound exists in enolimine form with monoclinic crystal lattice system and space group C2/c. Density functional calculation on L reveals that the enolimine form is more stable than the ketoamine form in gas phase. The DNA binding results suggest that the synthesized compound binds to DNA in a groove binding mode. The molecular docking studies demonstrate that L fits tightly into the spiral line of the DNA target in the minor groove.
Development of technology in the near past has stimulated the importance of discovering new materials, which find several applications. The aim of this work is to investigate the structural parameters and molecular docking studies on (E)-1-(4-Bromobenzylidene)Thiourea molecule (BBTU). The anti-bacterial proteins to identify the docking of ligand with protein. Based on this molecular design the effectiveness of the organic molecule was identified reasonably using computational techniques. In addition, the theoretical parameters such as bond parameters, vibrational assignments, NLO property, NBO analysis and global reactivity descriptors were calculated using DFT method at B3LYP/6-31G(d,p) basis set.
Experimental
Synthesis of (E)-1-(4-bromobenzylidene)Thiourea
Equimolar amount of 4-bromobenzaldehyde and thiourea were dissolved in 30 ml of absolute ethanol. The mixture was shaken to make homogenous solution. Few drops of catalyst acetic acid was added to increase the rate of reaction. The content was refluxed at 90°C for 3 hours. The completion of the reaction was monitored by thin layer chromatography. After the reaction was completed, the content was cooled the mixture was poured into water. The solid product obtained was filtered and purified using absolute ethanol.
Spectral Measurements
FT-IR and FT-Raman Spectra
The FT-IR spectrum of the synthesized BBTU was measured in the 4000–400 cm−1 region at the spectral resolution of 4 cm−1 using on SHIMADZU FT-IR affinity Spectrophotometer (KBr pellet technique) in Faculty of Marine Biology, Annamalai University, Parangipettai. The FT-Raman spectrum was recorded on BRUKER: RFS27 spectrometer operating at laser 100 mW in the spectral range of 4000–50 cm−1. FT-Raman spectral measurements were carried out from Sophisticated Analytical Instrument Facility (SAIF), Indian Institute of Technology (IIT), Chennai.

Melting point = 160˚, Yield = 85%
Computational Details
The entire calculations were performed at DFT/B3LYP/6-31G(d,p) level of basis set using Gaussian 03W 23 program package, invoking gradient geometry optimization 23, 24. The optimized structural parameters were used in the vibrational frequency calculations at the DFT level to characterize all the stationary points as minima. The vibrationally averaged nuclear positions of title molecule is used for harmonic vibrational frequency calculations resulting in IR and Raman frequencies together with intensities and Raman depolarization ratios. The vibrational modes were assigned on the basis of PED analysis using VEDA4 program 25. The Raman activity was calculated by using Gaussian 03W package and the activity was transformed into Raman intensity using Raint program 26 by the expression:
 (1)
(1)
Where Ii is the Raman intensity, RAi is the Raman scattering activities, νi is the wavenumber of the normal modes and ν0 denotes the wavenumber of the excitation laser 27.
Results and Discussion
Identification of Stable Conformer
After the determination of the low-energy structures belonging to the compound at HF level calculations. Further, the different positions of the atoms are surveyed by the detailed potential energy surface (PES) scans at B3LYP density functional method with 6-31G(d,p) basis set in the ground state to obtain the most stable conformer of the BBTU molecule. In this PES scan process, the potential energy surface is built by varying the C3‒C4‒C12‒N13 dihedral angle from 0˚-360˚ in every 10˚ rotation, while all the other geometrical parameters have been simultaneously relaxed. From Figure 1 the title compound with the planar conformation exhibits global minimum energy conformers at 0˚, 180˚ and 360˚ (-3388.47966 hartree), respectively. Whereas, the global maximum energy conformers were identified at 90˚ and 270˚ (-3388.465013 and -3388.465002 hartree) in PES. This is due to constrain appeared between the phenyl ring and thiourea moiety. In the stable conformer, there is no ring strain and planarity disturbance were observed in the title molecule. It is worth to discuss, that the theoretical findings supposed from this calculation level are more reliable and taken for further discussions. The energies of the various possible conformers of BBTU listed in the Table 1. The optimized structure of BBTU was shown in Figure 2.
Table 1. The various possible conformers of BBTU.| Rotation | Relative energy ( Hartree ) |
| 0 | -3388.47966 |
| 10 | -3388.479354 |
| 20 | -3388.478437 |
| 30 | -3388.476904 |
| 40 | -3388.474839 |
| 50 | -3388.472416 |
| 60 | -3388.469858 |
| 70 | -3388.467523 |
| 80 | -3388.465764 |
| 90 | -3388.465013 |
| 100 | -3388.465479 |
| 110 | -3388.467125 |
| 120 | -3388.469771 |
| 130 | -3388.472397 |
| 140 | -3388.474839 |
| 150 | -3388.476891 |
| 160 | -3388.478423 |
| 170 | -3388.479346 |
| 180 | -3388.47966 (min) |
| 190 | -3388.479347 |
| 200 | -3388.478423 |
| 210 | -3388.476891 |
| 220 | -3388.474839 |
| 230 | -3388.472397 |
| 240 | -3388.469771 |
| 250 | -3388.467349 |
| 260 | -3388.465621 |
| 270 | -3388.465002 |
| 280 | -3388.465614 |
| 290 | -3388.467283 |
| 300 | -3388.469858 |
| 310 | -3388.472416 |
| 320 | -3388.474839 |
| 330 | -3388.476904 |
| 340 | -3388.478437 |
| 350 | -3388.479355 |
Figure 1. The potential energy surface scan curve of BBTU.
Download figure
Figure 2. The optimized molecular structure of BBTU.
Download figure
Vibrational Assignments
The observed FT-IR, FT-Raman, computed harmonic wavenumber along with intensities and their assignments for the present compound are illustrated in Table 2. The comparison of simulated and recorded FT-IR and FT-Raman spectra were shown in Figure 3 and Figure 4, respectively. In general, harmonic frequencies overestimate the experimental values mainly due to anharmonicity and therefore empirical scaling procedures are used to compare the experimental and theoretical spectra. The theoretical vibrational wavenumber obtained for compound BBTU is interpreted by means of Potential energy distribution (PED %) calculations using VEDA4 software. The normal modes assignment of the theoretical frequencies is visualized and substantiated with the help of the GaussView 5.0 visualization program. The synthesized BBTU consists of 19 atoms and hence has 51 normal modes of vibrations. The molecule BBTU belongs to C1 symmetry.
Table 2. The fundamental vibrational assignments of BBTU.| Mode No | Theoretical Wavenumber (cm -1 ) | Experimental Wavenumber (cm -1 ) | IR Int. | Raman Int. | PED≥10% | ||
| Un Scaled | Scale d | FT- IR | FT-Rama n | ||||
| 1 | 3721 | 3579 | 21.27 | 0.70 | νN14H18(100)+ νN14H19(100) | ||
| 2 | 3575 | 3439 | 3419m | 26.95 | 1.61 | νN14H18(100)+ νN14H19(100) | |
| 3 | 3209 | 3087 | 0.56 | 2.08 | νC2H7(96)+ νC3H8(97) | ||
| 4 | 3206 | 3084 | 0.57 | 1.07 | νC6H10(89) | ||
| 5 | 3196 | 3074 | 0.01 | 0.58 | νC2H7(96)+ νC3H8(97) | ||
| 6 | 3174 | 3054 | 3062vs | 0.98 | 0.73 | νC5H9(94) | |
| 7 | 3047 | 2931 | 2922s | 1.63 | 0.30 | νC12H17(100) | |
| 8 | 1675 | 1611 | 1658vs | 1636w | 98.12 | 97.31 | νN13C12(72) |
| 9 | 1628 | 1566 | 39.43 | 100.00 | νC2C3(48)+ νC6C5(54)+βC3C4C5(46)+ βH18N14H19(88) | ||
| 10 | 1619 | 1558 | 117.80 | 31.72 | νN14C15(70)+ βH18N14H19(88) | ||
| 11 | 1597 | 1536 | 31.93 | 24.06 | νC2C1(57)+ νC4C3(77)+ βC6C5C4(49) | ||
| 12 | 1517 | 1459 | 1433s | 12.04 | 1.64 | βH7C2C3(64)+ βH8C3C2(65)+ βH9C5C6(66)+ βH10C6C5(57) | |
| 13 | 1431 | 1377 | 1373s | 1383m | 11.95 | 5.41 | νC2C3(48)+ νC6C5(54) |
| 14 | 1413 | 1360 | 44.47 | 1.04 | νN14C15(70)+ βH18N14H19(88)+ βH17C12N13(73) | ||
| 15 | 1376 | 1324 | 1334s | 19.84 | 0.19 | νN14C15(70)+ βH17C12N13(73) | |
| 16 | 1326 | 1276 | 1278vs | 9.47 | 7.11 | νC1C6(61)+ νC4C3(77)+ βH7C2C3(64) | |
| 17 | 1320 | 1270 | 0.73 | 0.54 | νC1C6(61)+ νC2C1(57)+ βH7C2C3(64)+ βH8C3C2(65)+βH9C5C6(66)+ βH10C6C5(57) | ||
| 18 | 1288 | 1239 | 1207s | 100.00 | 46.84 | νN13C15(69)+ βH18N14C15(81) | |
| 19 | 1241 | 1194 | 48.13 | 75.46 | βC3C4C5(46)+ νC12C4(59)+ βH18N14C15(81)+ βH17C12N13(73) | ||
| 20 | 1193 | 1148 | 1166s | 17.72 | 41.22 | βH8C3C2(65)+ βH9C5C6(66)+ βH10C6C5(57) | |
| 21 | 1129 | 1086 | 1093s | 1.50 | 0.80 | νC2C3(48)+ νC6C5(54)+ βH7C2C3(64)+ βH8C3C2(65)+βH9C5C6(66)+ βH10C6C5(57) | |
| 22 | 1080 | 1039 | 1028w | 18.95 | 13.14 | νC1C6(61)+ νC2C1(57)+ νBr11C1(90) | |
| 23 | 1049 | 1009 | 1.55 | 0.48 | ГH17C12N13C15(83) | ||
| 24 | 1026 | 987 | 14.98 | 2.57 | βC3C4C5(46)+ βC6C5C4(49)+ βC4C3C2(74) | ||
| 25 | 996 | 958 | 0.00 | 0.02 | ГH7C2C3C4(87)+τC3C2C4H8(87) | ||
| 26 | 991 | 953 | 70.81 | 73.11 | νN13C15(69)+ βH18N14C15(81) | ||
| 27 | 972 | 935 | 933s | 0.01 | 0.09 | τC5C4C6H9(88)+ ГH10C6C5C4(88) | |
| 28 | 874 | 841 | 6.07 | 2.31 | νC4C3(77)+ νC12C4(59)+ βN13C12C4(44)+ βC2C1C6(59) | ||
| 29 | 848 | 815 | 812vw | 6.51 | 0.21 | ГH7C2C3C4(87)+ τC3C2C4H8(87) | |
| 30 | 838 | 806 | 7.03 | 0.07 | τC5C4C6H9(88)+ ГH10C6C5C4(88) νS16C15(67) | ||
| 31 | 779 | 750 | 740vs | 733vs | 6.06 | 19.36 | ГC1C6C4C5(84)+ ГC1C6C2C3(82)+ ГC3C2C4C5(69) |
| 32 | 728 | 700 | 702s | 0.76 | 0.02 | νBr11C1(90)+ βC2C1C6(59)+ βC15N13C12(72) | |
| 33 | 685 | 659 | 1.77 | 6.58 | ГC4C12N13C15(71)+ τS16N13N14C15(72) | ||
| 34 | 663 | 638 | 0.00 | 1.08 | νC4C3(77)+ βC4C3C2(74) | ||
| 35 | 643 | 619 | 0.16 | 2.08 | ГH18N14C15S16(98)+ τN14H18C15H19(94) | ||
| 36 | 621 | 598 | 565 | 0.53 | 0.07 | νS16C15(67)+ βN14C15N13(63)+ βC5C4C12(72)+βC15N13C12(72) | |
| 37 | 518 | 498 | 514 | 1.23 | 3.98 | ГC1C6C2C3(82)+ ГC3C2C4C5(69) | |
| 38 | 505 | 486 | 478s | 10.75 | 0.46 | νC12C4(59)+ νBr11C1(90)+ βN14C15S16(83) | |
| 39 | 487 | 468 | 449 | 0.52 | 2.00 | ГC1C6C4C5(84)+ ГC1C6C2C3(82) | |
| 40 | 420 | 404 | 405 | 0.23 | 0.04 | νBr11C1(90)+ βN14C15N13(63)+ βC5C4C12(72)+βN14C15S16(83) | |
| 41 | 380 | 365 | 1.21 | 1.62 | ГH18N14C15S16(98)+ τN14H18C15H19(94) | ||
| 42 | 371 | 357 | 59.73 | 0.53 | ГC3C2C4C5(69)+ ГC3C2C1Br11(91)+ ГC6C5C4C12(81) | ||
| 43 | 332 | 319 | 0.73 | 2.65 | νBr11C1(90)+ βC2C1C6(59)+ βN14C15N13(63)+ βC5C4C12(72)+ βC6C1Br11(70) | ||
| 44 | 310 | 298 | 4.48 | 0.77 | βC6C1Br11(70) | ||
| 45 | 226 | 218 | 0.78 | 0.43 | νC12C4(59)+ νBr11C1(90)+ βC15N13C12(72)+ βN14C15S16(83) | ||
| 46 | 169 | 163 | 0.05 | 3.61 | ГC3C4C12N13(78)+ ГC3C2C1Br11(91) | ||
| 47 | 161 | 155 | 3.53 | 0.33 | ГC4C12N13C15(71)+ ГC3C2C1Br11(91) | ||
| 48 | 149 | 144 | 0.90 | 0.09 | βN13C12C4(44)+ βC5C4C12(72)+ βC15N13C12(72) | ||
| 49 | 74 | 71 | 0.34 | 1.57 | ГC3C4C12N13(78)+ ГC4C12N13C15(71)+ ГC3C2C1Br11(91)+ ГC6C5C4C12(81) | ||
| 50 | 46 | 44 | 0.02 | 1.12 | ГN14C15N13C12(83) | ||
| 51 | 5 | 5 | 2.86 | 22.72 | νN13C15(69)+ βH18N14C15(81) | ||
Figure 3. The experimental and theoretical FT-IR spectra of BBTU.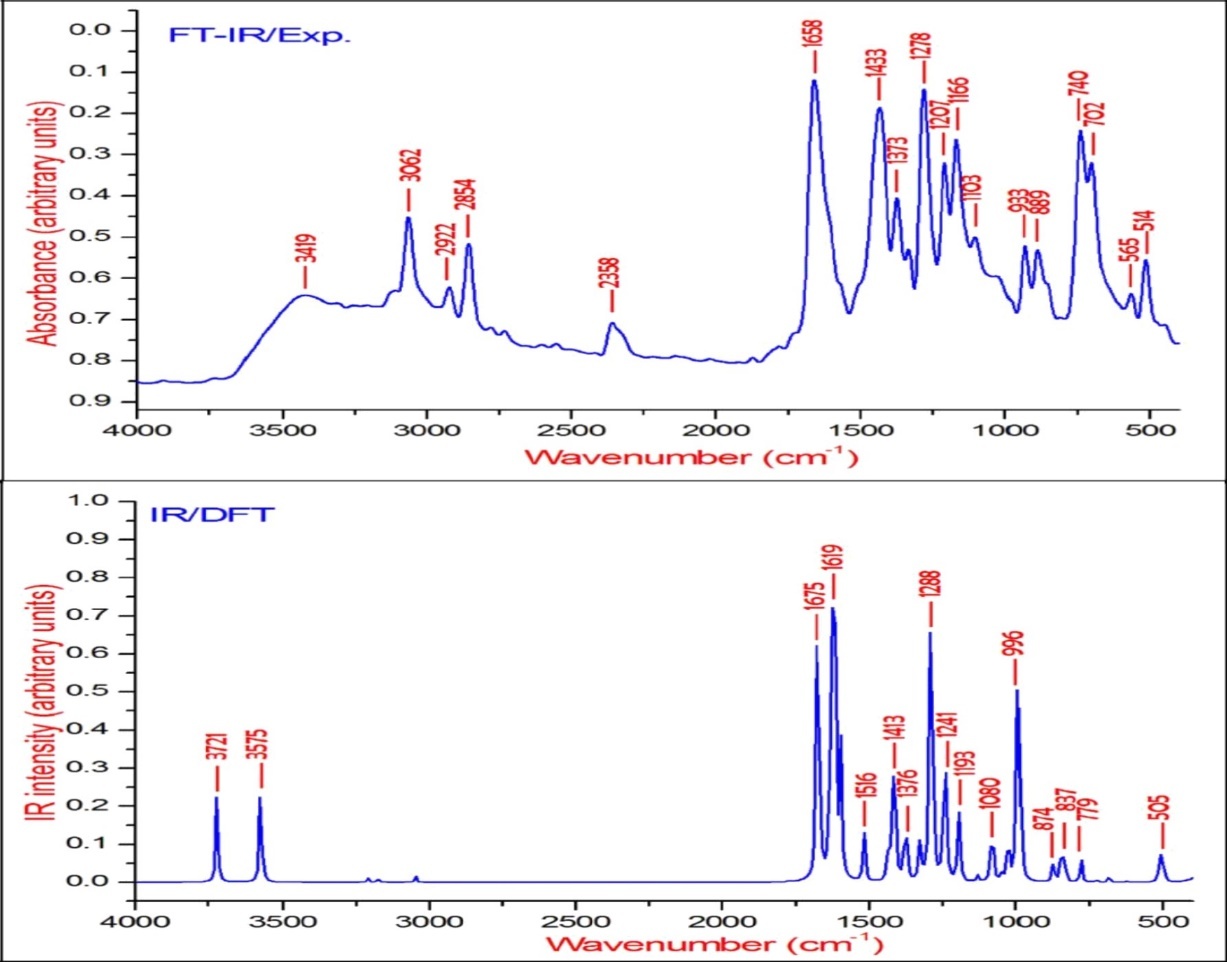
Download figure
Figure 4. The experimental and theoretical FT-Raman spectra of BBTU.
Download figure
N-H Vibrations
The νN-H stretching vibrations normally appear in the range of 3500-3300 cm-128. The experimental wavenumber of 3419 cm-1 in the FT-IR spectrum of BBTU is assigned for N-H stretching vibration. The theoretical N-H stretching wavenumber are visualized at 3579 cm-1 for asymmetric and at 3439 cm-1 for symmetric stretching wavenumber. Both the theoretical and experimental vibrational frequencies are pure vibrations with PED of 100%. The N-H scissoring vibrational mode appears in the 1638-1575 cm–1 region with strong to very strong IR intensity, but exhibits weak or no Raman scattering. The NH2 scissoring deformation is calculated at 1566 cm–1 and the NH2 wagging mode is calculated at 357 cm–1.
C-Br Vibrations
Bromine compounds absorb strongly in the region 650-485 cm-1 due to the C-Br stretching vibrations 29. The theoretical wavenumbers of C–Br stretching vibration coupled with other group vibrations. According to PED, there is no pure C–Br band vibration. In our present study, the C-Br vibration was calculated at 659 cm-1 along with CCC and CNC bending vibrations. The 90% of PED contribution confirms the corresponding wavenumber. The C–Br out-of-plane bending and in-plane bending vibrations are assigned to the Raman bands at 298 and 218 cm−1, respectively.
C=S Vibrations
The C=S is less polar than the C=O group and encountered great difficulty due to vibrational mixings between C=S stretching the other vibrational modes. The C=S group is found over the wide range of 1035-245 cm–1 whereas, C–S stretching vibration results strong bands in Raman spectra which are normally easy to identify and is difficult in Infrared region 29. In consequence, the band is not intense, and it falls at lower frequencies, where it is much more susceptible to coupling effects and identification is therefore difficult and uncertain 30. In our present study the C=S band is observed in FT-IR at 740 cm–1 and in FT-Raman spectrum at 733 cm–1 as strong bands. Its theoretical wavenumber calculated at 750 cm–1 with 67% of PED contribution.
C-N Vibrations
The C=N stretching skeletal bands are observed in the range 1650–1550 cm–131. For BBTU, the C=N observed at 1658 cm–1 in FT-IR spectrum and at 1636 cm–1 in FT-Raman spectrum. The DFT prediction for the corresponding mode is calculated at 1611 cm–1 with 72% contribution in PED. The C-N stretching vibrations are observed in combination with other vibration modes and are dispersed in the range of 1062-1407 cm–1 32. In this study, the C15-N14 and C15-N13 were observed at 1334 and 1207 cm–1 in FT-IR spectrum, besides with bending vibrations of βCNH. The corresponding theoretical wavenumber for C-N vibrations are calculated at 1324 and 1239 cm–1.
Aromatic Ring Vibrations
The aromatic ring structure shows the presence of C−H stretching vibrations in the region 3100–3000 cm–1, which is the characteristic region of C−H stretching vibrations 33. The wavenumbers computed in the region 3087–3054 cm–1 is assigned to C−H stretching vibrations of BBTU. The strong band observed at 3062 cm–1 in FT-IR spectrum was assigned to C−H stretching vibrations of the phenyl ring. From Table 2, it is evident that the calculated wavenumber at 3087, 3084, 3074 and 3054 cm–1 are pure modes, they almost contributing more than 90% of PED. The C−H in-plane ring bending vibrations are normally occurs as a number of strong to weak intensity bands in the region 1000–1300 cm–134. The bands observed at 1166 cm–1 in FT-IR spectrum and at 1093 cm–1 inFT-Raman spectrum are assigned for the C−H in-plane bending vibrations of the aromatic ring. The theoretical wavenumber at 1320, 1193 and 1129 cm–1 are in good agreement with the experimental values. The C−H out-of-plane bending vibration is expected in the range 750–1000 cm–1 in the FT-IR spectra of substituted benzenes 35. The position of C−H out-of-plane vibration is determined almost extensively by the relative position of the substituents and is independent of nature 36. The vibrational bands observed at 933 and 812 cm–1 in FT-IR spectrum were assigned for the C−H out-of-plane bending vibrations of the phenyl ring.
The ring stretching vibrations are very much important in the spectrum of aromatic compounds and are highly characteristic of the aromatic ring itself. In general, the C−C bands are of variable intensity and observed at 1625–1590, 1590–1575, 1540–1470, 1460–1430 and1380–1280 cm–1 from the frequency ranges given by Varsanyi for the five bands in the finger print region 36. In BBTU, the strong vibrational bands at 1433, 1373, 1278 cm–1 in FT-IR spectrum and at 1383 cm–1 in FT-Raman spectrum were assigned for C-C vibrations of the phenyl ring. The C-C theoretical wavenumber at 1536, 1459, 1377 and 1276 cm–1 have shown good agreement with the experimental wavenumber. The phenyl ring breathing mode and trigonal bending vibrations are calculated at 1039 and 987 cm–1. The weak band at 1028 cm–1 assigned to the phenyl ring breathing mode vibration.
NBO Analysis
The natural bond orbital (NBO) calculations were performed in order to investigate the intra- and inter-molecular bonding interactions among bonds in BBTU. The intra-molecular charge transfer and hyperconjugative interactions of BBTU were also analyzed. The NBO analysis gives information about energy of the interactions between filled Lewis type NBOs and empty non-Lewis type NBOs and estimating their energy by second order perturbation theory. A filled bonding or lone pair orbital can act as a donor and a vacant anti-bonding or lone pair orbital can act as an acceptor. These intra-molecular interactions between orbitals can strengthen and weaken bonds in the molecular system. The interaction between a lone pair donor and anti-bonding acceptor orbital will weaken the bond associated with the anti-bonding orbital. In contrast, the interaction with a bonding pair as the acceptor orbital will strengthen the bond 37, 38, 39. The second order Fock matrix was performed to evaluate the donor (i) – acceptor (j) interactions in the NBO basis. The stabilization energies of the title compound have been investigated using second order perturbation theory. The stabilization energy E(2) associated with each donor NBO(i) and acceptor NBO(j) delocalization is calculated from the second order perturbation approach 40 by the following equation.
 (2)
(2)
Where, qi is the donor orbital occupancy, εi and εj are diagonal elements (orbital energies) and F(i, j) is off diagonal NBO Fock matrix elements.
NBO analysis was performed by the B3LYP level of theory with 6-31G(d,p) basis set for the molecule BBTU. NBO theory allows the assignment of hybridization of atomic lone pairs and of the atoms involved in bond orbitals. These are important data in spectral interpretation as the frequency ordering is related to the bond hybrid composition. The second order perturbation energy values E(2) collected in Table 3 reveals the important interactions between the Lewis and non-Lewis type NBO orbitals of BBTU molecular system consisting para bromo-substituted phenyl ring fused with thiourea. In the studied molecule, the strong intra-molecular hyperconjugative interactions were observed between lone pair electrons of N, S with σ and π anti-bonding electrons with C‒C, C‒N and C‒S resulting in intra-molecular charge transfer (ICT) causing stabilization of the BBTU molecular system. Enormous stabilization energy of 332.17 KJ/mol was found for LP(1)N14→C15-S16 interactions, this larger stabilization show the larger delocalization of electrons in the title molecule. Lone pair electrons form S16 distributed stabilization energy to σ* (N13‒C15), (N14‒C15) leads to the stabilization of 52.93, 46.36 KJ/mol for BBTU. The magnitude of charges transferred from bonding lone pair of N13 with ED (1.9041) to σ* (C12‒H17), (C15‒S16) shows 43.43 and 46.19 KJ/mol stabilization energy. The π(C12‒ N13)→π*(C4‒C5), (C15‒S16) leads to the stabilization of system with energy of 34.48 KJ/mol to phenyl ring system and 98.99 KJ/mol to C=S of the thiourea moiety.
Table 3. The second order perturbation energies and interactions of BBTU| Type | Donor | ED(e) | Acceptor | ED(e) | E 2 KJ/ mol | E 2 Kcal/ mol |
| σ-σ* | C1-C2 | 1.98023 | C1-C6 | 0.02771 | 14.48 | 3.46 |
| C2-C3 | 0.01683 | 14.39 | 3.44 | |||
| σ-σ* | C1-C6 | 1.98017 | C1-C2 | 0.02818 | 14.39 | 3.44 |
| C5-C6 | 0.01668 | 14.52 | 3.47 | |||
| π-π* | C1-C6 | 1.66594 | C2-C3 | 0.27013 | 72.38 | 17.3 |
| C4-C5 | 0.37576 | 83.18 | 19.88 | |||
| σ-σ* | C1-Br11 | 1.98234 | C2-C3 | 0.01683 | 12.68 | 3.03 |
| C5-C6 | 0.01668 | 12.89 | 3.08 | |||
| π-π* | C2-C3 | 1.66262 | C1-C6 | 0.37698 | 93.26 | 22.29 |
| C4-C5 | 0.37576 | 77.7 | 18.57 | |||
| σ-σ* | C3-H8 | 1.97689 | C1-C2 | 0.02818 | 17.07 | 4.08 |
| C4-C5 | 0.02285 | 19.25 | 4.6 | |||
| π -π* | C4-C5 | 1.61775 | C1-C6 | 0.37698 | 87.95 | 21.02 |
| C2-C3 | 0.27013 | 82.84 | 19.8 | |||
| C12-N13 | 0.13717 | 86.23 | 20.61 | |||
| π-π* | C12-N13 | 1.86787 | C4-C5 | 0.37576 | 34.48 | 8.24 |
| C15-S16 | 0.42547 | 98.99 | 23.66 | |||
| σ-σ* | C12-H17 | 1.98519 | C3-C4 | 0.02677 | 17.61 | 4.21 |
| σ-σ* | N13-C15 | 1.97971 | C4-C12 | 0.03174 | 16.99 | 4.06 |
| C12-N13 | 0.01022 | 5.15 | 1.23 | |||
| N14-H18 | 0.00883 | 12.34 | 2.95 | |||
| σ-σ* | N14-C15 | 1.99299 | C12-N13 | 0.01022 | 11.05 | 2.64 |
| σ-σ* | N14-H18 | 1.99025 | N13-C15 | 0.06856 | 13.77 | 3.29 |
| σ-σ* | N14-H19 | 1.987 | C15-S16 | 0.0345 | 22.38 | 5.35 |
| π-π* | C15-S16 | 1.97717 | C12-N13 | 0.13717 | 14.81 | 3.54 |
| C15-S16 | 0.42547 | 15.61 | 3.73 | |||
| n-σ* | LP(1)Br11 | 1.99344 | C1-C2 | 0.02818 | 6.44 | 1.54 |
| C1-C6 | 0.02771 | 6.49 | 1.55 | |||
| n-σ* | LP(2)Br11 | 1.97494 | C1-C2 | 0.02818 | 14.43 | 3.45 |
| C1-C6 | 0.02771 | 14.52 | 3.47 | |||
| n-π* | LP(3)Br11 | 1.92396 | C1-C6 | 0.37698 | 43.51 | 10.4 |
| n-σ* | LP(1) N13 | 1.90412 | C4-C12 | 0.03174 | 8.49 | 2.03 |
| C12-H17 | 0.04197 | 43.43 | 10.38 | |||
| N14-C15 | 0.05274 | 16.15 | 3.86 | |||
| C15-S16 | 0.0345 | 46.19 | 11.04 | |||
| n-π* | LP(1) N14 | 1.6631 | C15-S16 | 0.42547 | 332.17 | 79.39 |
| n-σ* | LP(1) S16 | 1.98536 | N13-C15 | 0.06856 | 15.19 | 3.63 |
| N14-C15 | 0.05274 | 10.38 | 2.48 | |||
| n-σ* | LP(2) S16 | 1.87959 | C12-H17 | 0.04197 | 5.69 | 1.36 |
| N13-C15 | 0.06856 | 52.93 | 12.65 | |||
| N14-C15 | 0.05274 | 46.36 | 11.08 |
The electron density of the six conjugated double bonds of the phenyl ring (~1.66e) clearly demonstrates the strong delocalization through the interactions π(C1‒C6)→π*(C2‒ C3), (C4‒C5), π(C2‒C3)→π*(C1‒C6), (C4‒C5) and π(C4‒C5)→π*(C1‒C6), (C2‒C3) with the stabilization energies of about 72.38, 83.18, 93.26, 77.7 and 87.95, 82.84 KJ/mol, respectively. The donor LP(3) Br11→ π*(C1‒C6) transfers the hyperconjugative interaction energy of about 43.51 KJ/mol to acceptor anti-bond. The bonding π(C4‒C5)→anti-bonding π*(C12‒N13) interaction leads to the stabilization of 86.23 KJ/mol to the thiourea side chain of the molecular system. These types of intra-molecular stability of BBTU are responsible for pharmaceutical and biological properties. Hence, the BBTU structure is stabilized by these orbital interactions.
Non-Linear Optics
The first order hyperpolarizabilities (β0, α0 and Δα) of BBTU is calculated using DFT/B3LYP/6-31G(d,p) basis set, based on the finite-field approach. In the presence of an applied electric field, the energy of a system is a function of the electric field. First hyperpolarizability is a third rank tensor that can be described by a 3x3x3 matrix. The 27 components of the 3D matrix can be reduced to 10 components due to Kleinman symmetry 41. It can be given in the lower tetrahedral format. It is obvious that the lower part of the 3x3x3 matrix is a tetrahedral. The components of β are defined as the coefficients in the Taylor series expansion of the energy in the external electric field. When the external electric field is weak and homogeneous, this expansion becomes:
 (3)
(3)
Where E0 is the energy of the unperturbed molecules, Fα is the field at the origin, and µα, ααβand βαβγ are the components of the dipole moment, polarizability and the first hyperpolarizabilities, respectively. The total static dipole moment μ, the mean polarizability α0, the anisotropy of polarizability Δα and the mean first hyperpolarizability β0, using the x, y, z components 42 are defined as
 (4)
(4)
 (5)
(5) (6)
(6)
 (7)
(7)
Many organic molecules, containing conjugated π electrons are characterized by large values of molecular first hyperpolarizabilities, were analyzed by means of vibrational spectroscopy 43, 44, 45, 46. The intra molecular charge transfer from the donor to acceptor group through a single-double bond conjugated path can induce large variations of both the molecular dipole moment and the molecular polarizability, making IR and Raman activity strong at the same time 47.
Theoretical investigation plays an important role in understanding the structure – property relationship, which is able to assist in designing novel NLO materials. It is well known that the higher values of dipole moment, molecular polarizability and hyperpolarizability are important for more active NLO properties. It is evident from Table 4, the molecular dipole moment (μ), molecular polarizability and hyperpolarizability are calculated about 1.2315 (D), 3.78 and 23.70 x10-30esu, respectively. The β0 value of the title compound is sixtyfour times greater than that of urea. Hence, our title molecule is an interesting object for Non-linear Optics.
Table 4. The Non-Linear Optical properties of BBTU.| Parameters | B3LYP/6-31G(d,p) |
|---|---|
| Dipole moment ( μ ) | Debye |
| μx | -0.6392 |
| μy | 1.0526 |
| μz | 0.0005 |
| μ | 1.2315 Debye |
| Hyperpolarizability ( β0) | x10-30esu |
| βxxx | 2912.47 |
| βxxy | 29.87 |
| βxyy | -159.94 |
| βyyy | -15.77 |
| βxxz | 0.07 |
| βxyz | 0.01 |
| βyyz | 0.03 |
| βxzz | -8.98 |
| βyzz | 3.63 |
| βzzz | 0.31 |
| β0 | 23.70 X10 -30 esu |
Energy Gap Analysis
Frontier molecular orbitals are nothing but, the highest occupied molecular orbitals and lowest unoccupied molecular orbitals of the molecule. The LUMO as an electron acceptor represents the ability to obtain an electron; donor represents the ability to donate an electron. The energy gap of HOMO-LUMO explains the eventual charge transfer interaction within the molecule, which influences the biological activity of the molecule. The positive and negative phases are represented in red and green colour respectively. The energy of the two important FMOs such as the highest occupied molecular orbitals (HOMO), the lowest unoccupied molecular orbitals (LUMO) have been calculated. The frontier molecular orbitals of the title molecule are mapped in Figure 5.
Figure 5. The frontier molecular orbitals of BBTU
Download figure
As seen from Figure 5, the HOMO is localized over thiourea side chain. LUMO is localized over entire region of the title molecule. The analysis of the wave function indicates that the electron absorption corresponds to the transition from the ground to the first excited state and is mainly described by one electron excitation from the HOMO to LUMO. The HOMO→LUMO transition implied that an electron density transfer from thiourea to bromo substituted phenyl ring. The values of the energy separation between the HOMO→LUMO is identified as 3.2860 eV. This HOMO–LUMO gap leads to high excitation energies, a good stability for BBTU. In addition, it is noted that there is an overlap between the HOMO and LUMO, which is necessary for obtaining large second-order response 49. The energies of the FMOs are used for the determination of global reactivity descriptors. The frontier molecular orbital energies are tabulated in the Table 5.
Table 5. The frontier molecular orbital energies of BBTU| Orbitals | Energies a.u | Energies eV | K.E |
| 56O | -0.290816 | -7.913394 | 1.863647 |
| 57O | -0.289536 | -7.878564 | 1.139333 |
| 58O | -0.263996 | -7.183595 | 1.829087 |
| 59O | -0.233738 | -6.360245 | 1.775288 |
| 60O | -0.221343 | -6.022964 | 1.830617 |
| 61V | -0.10058 | -2.736882 | 1.556817 |
| 62V | -0.040214 | -1.094263 | 1.353532 |
| 63V | -0.02221 | -0.604356 | 2.275447 |
| 64V | -0.021277 | -0.578968 | 1.633853 |
| 65V | 0.035886 | 0.9764939 | 0.852772 |
Molecular Electrostatic Potential
MEP and electrostatic potential are useful quantities to illustrate the charge distributions of molecules and used to visualize variably charged regions of a molecule. Therefore, the charge distributions can give information about how the molecules interact with another molecule. The reactive behavior of the BBTU molecule is visualized with the help of three dimensional MEP surface. The molecular electrostatic potential and the contour map of the BBTU are shown in Figure 6. The MEP surface of the molecule under investigation is constructed by using B3LYP/ 6-31G(d,p) method. MEP surface describes the charge distribution in the molecule and helps in predicting the sites for nucleophilic and electrophilic attack in the molecule. The region of negative charge is pictured out by red color, which indicates the electrophilic attack sites of our molecule. The red region localized over the ‒CH=N imine linkage and in core region of phenyl ring of the title molecule. The region of positive charge is pictured out by blue color, which indicates the nucleophilic attack sites were localized over NH2 group of the thiourea moiety and around bromine atom attached with phenyl ring of the BBTU. The green color corresponds to a potential half way between the red and blue regions, also represents the neutral charge. The contour map of BBTU describes the electron density mapped surface of the BBTU.
Figure 6. The MEP mapped surface and contour map of BBTU.
Download figure
Global Reactivity Descriptors
According to Koopmans theorem 50, the first ionization energy of the molecular system is equal to the negative of the orbital energy of the HOMO and electron affinity as the energy of the LUMO of respective system. In the framework of energy gap analysis, the ionization energy and electron affinity can be expressed as I = −EHOMO and A = −ELUMO by HOMO-LUMO orbital energies. The combination of electron affinity and ionization energy gives the electronic chemical potential, μ = 1/2(EHOMO+ELUMO). Global hardness expressed by η = 1/2(ELUMO-EHOMO), global electrophilicity index by ω = μ2/2η and softness s = 1/η 51. Chemical potential and hardness are the better descriptor of global chemical reactivity. For the title compound, EHOMO = −6.022964 eV, ELUMO = −2.736882 eV, Energy gap = HOMO– LUMO = 3.286082 eV, Ionization potential I = −6.022964, Electron affinity A = −2.736882 global hardness η = 1.643041, chemical potential μ = 4.379923, global electrophilicity ω = μ2/2η = 5.83787182eV and softness ζ=1/η = 0.60862754. The electrophilicity indexmeasures the stabilization in energy when the system acquires an additional electronic charge from the environment. Electrophilicity encloses both the ability of an electrophile to gain additional electronic charge and the resistance of the system to exchange electronic charge with the environment.
Molecular Docking Studies
Protein–ligand interactions play a critical role in the distribution, metabolism and transport of small molecules in biological systems and processes 52. AutoDock is a suite of automated docking tools designed to predict how small molecules, such as substrates or drug candidates, bind to a receptor of known three-dimensional structure. With the aim to investigate the binding mode, molecular modeling study was performed using AutoDock Tools for docking 53. 5NI was chosen to be docked into the active site of different receptors 1JIJ and 3U2D of antimicrobial proteins which was obtained from Protein Data Bank (PDB). Ligands were docked into the functional sites of the respective proteins individually and the docking energy was monitored to achieve a minimum value. AutoDock results indicate the binding position and bound conformation of the peptide, together with a rough estimate of its interaction. Docked conformation which had the minimum binding energy was selected to analyze the mode of binding. The Lamarckian genetic algorithm 54 was applied to search for protein-ligand interaction with and for visualization performed using PyMOL 55.
The objective of present study was to evaluate the drug activity and binding affinity of BBTU with a target protein using the program of AutoDock. The resulting free energy of binding and inhibition constant of BBTU with protein 3U2D and IJIJ observed at the end of docking simulation is shown in Table 6. Pictorial representations of best possible binding sites of ampicillin and BBTU with 3U2D receptor were shown in Figure 7. From Figure 8 it is shown that BBTU ligand was focused in the groove of the protein and hydrogen bonding interactions are highlighted. The BBTU was docked deeply with the binding pocket of 3U2D forming two hydrogen bonds with SER55 and ILE51. The NH2 group of BBTU forms hydrogen bonding with the distances of 2.1 and 2.2 Å with oxygen atoms of the residues SER55 and ILE51, respectively. The best binding score of BBTU was obtained as -5.53kcal/mol and the estimated inhibition constant of BBTU was found to be 87.90 μM against DNA gyrase. Hydrophobic and van der Waals interactions were also detected in the lead molecule of BBTU with DNA gyrase are with the residues of PRO87, ILE 175, VAL52, GLY83 and GLY85. The pi-anion and pi-alkyl interactions are identified with the residues of GLU58 and ILE86 from the 2D model interactions. These type of π-interactions are due to the electron density in the core of the phenyl ring are clearly demonstrated by the contour map of the BBTU. This result indicates the antibacterial activity of the title compound.
Table 6. Free binding energy and estimated inhibition constants of BBTU with the target protein.| Ligand | T arget protein (Receptor) | Binding energy [Kcal/ Mol ] | Estimatedinhibitionconstant(μM) | Reference RMSD |
| BBTU | 3U2D | -5.53 | 87.9 | 25.18 |
| BBTU | 1JIJ | -4.53 | 481.2 | 89.6 |
| Ligand | Protein [ PDB ID] | Protein bounded residues | ||
| 3U2D | ILE51, SER55, VAL79, THR173, | |||
| BBTU | ASP81, ILE86, GLU58, ARG84 | |||
| ASN54, VAL52, GLY83, GLY85 | ||||
| PRO87, ILE 175 | ||||
Figure 7. The Binding pose of Ampicillin (a) and BBTU (b) with 3U2D receptor.
Download figure
Figure 8. The binding poses and hydrogen bonding interactions of BBTU with 3U2D.
Download figure
Conclusion
The organic molecule (E)-1-(4-bromobenzylidene)thiourea was synthesized and characterized using FT-IR and FT-Raman spectral techniques. The optimized stable conformer of the title compound was analyzed by the potential energy surface scan. Further the stable conformer is used to investigate it properties theoretically using DFT method. The fundamental vibrational assignments are characterized by FT-IR and FT-Raman spectral data. More precisely assigned with the help of potential energy distributions (PED). The stability arising due to the intra-molecular interactions are identified from the NBO analysis. From NBO results, the stabilization are arise by the lone pair interactions of oxygen and nitrogen atoms. The charge sites are identified by mapping molecular electrostatic potential of BBTU. The nonlinear optical property of the BBTU was sixty four times greater than the reference Urea. It shows that, the BBTU molecule is suitable for the optical functioning. A molecular docking result exhibits the anti-bacterial activity of the title molecule.
References
- 3.Mounika K, Anupama B, Pragathi J, Gyanakumari C. (2010) Synthesis¸ Characterization and Biological Activity of a Schiff Base Derived from 3-Ethoxy Salicylaldehyde and 2-Amino Benzoic acid and its Transition Metal Complexes. , J. Sci. Res 3, 513-524.
- 4.Venkatesh P. (2011) Synthesis, characterization and antimicrobial activity of various schiff bases complexes of Zn (II) and Cu (II) ions. , Asian J. Pharma. Health Sci 1, 8-11.
- 5.Yıldız M, Dülger B, S Y Koyuncu, B M Yapıcı. (2004) Synthesis and antimicrobial activity of bis(imido) Schiff bases derived from thiosemicarbazide with some 2-hydroxyaldehydes and metal complexes. , J. Ind. Chem. Soc 81, 7-12.
- 6.Ünver H, Yıldız M, Dülger B, Özgen Ö, Kendi E et al. (2005) Spectroscopic studies, antimicrobial activities and crystal structures of N-(2-hydroxy-3-methoxybenzalidene)1-aminonaphthalene. , J. Mol. Struct 737, 159-164.
- 7.Yıldız M, Ünver H, Dülger B, Erdener D, Ocak N et al. (2005) Spectroscopic study, antimicrobial activity and crystal structures of N-(2-hydroxy-5-nitrobenzalidene) 4-aminomorpholine and N-(2-hydroxy-1-naphthylidene)4-aminomorpholine. , J. Mol. Struct 738, 253-260.
- 8.Yıldız M, Dülger B, Çınar A. (2005) Synthesis and characterization of new crown ethers of schiff base type and their complexes. , J. Ind. Chem. Soc 82, 414-420.
- 10.Kiraz A, Yıldız M, Dülger B. (2009) Synthesis and Characterization of Crown Ethers. , Asian J. Chem 21(6), 4495-4507.
- 11.Sondhi S M, Singh N, Kumar A, Lozach O, Meijer L. (2006) Synthesis, anti-inflammatory, analgesic and kinase (CDK-1, CDK-5 and GSK-3) inhibition activity evaluation of benzimidazole/benzoxazole derivatives and some Schiff’s bases. , Bioorg. Med. Chem 14(11), 3758-3765.
- 12.P G Cozzi. (2004) Metal–Salen Schiff base complexes in catalysis: practical aspects. , Chem. Soc. Rev 410-421.
- 13.Chandra S, Sangeetika J. (2004) EPR and electronic spectral studies on copper(II) complexes of some N-O donor ligands. , J. Indian Chem. Soc 81, 203-206.
- 14.P G Avaji, Kumar C H V, S A Patil, K N Shivananda, Nagaraju C. (2009) Synthesis, spectral characterization, in-vitromicrobiological evaluation and cytotoxic activities of novel macrocyclic bis hydrazone. , Eur. J. Med. Chem 44(9), 3552-3559.
- 15.Dalapati S, Jana S, Guchhait N. (2014) Anion recognition by simple chromogenic and chromo-fluorogenic salicylidene Schiff base or reduced-Schiff base receptors. , Spect. Acta Part A: Mol. Biomol. Spect.129 499-508.
- 16.K S Saravana, Shilpa B, K S Suban, Kumar S K A. (2014) Fluoride selective colorimetric sensor based on cefetamet pivoxil drug. , J. Fluorine Chem 164, 51-57.
- 17.Sharma D, Ashok Kumar SK, Sahoo S K. (2014) Vitamin B6 cofactor derived chemosensor for the selective colorimetric detection of acetate anions. , Tetrahedron Lett 55(4), 927-930.
- 18.Huang X, He Y, Chen Z, Hu C. (2009) Colorimetric Sensors for Anion Recognition Based on the Proton Transfer Signaling Mechanism. , Chin. J. Chem 27, 1526-1530.
- 20.Al-Sehemi A G. (2012) Synthesis, characterization and DFT study of methoxybenzylidene containing chromophores for DSSC materials. , Spectrochimica Acta Part A 91, 239-243.
- 21.Wesley Jeevadason A, Kalidasa Murugavel K, Neelakantan M A. (2014) Review on Schiff bases and their metal complexes as organic photovoltaic materials, Renewable and Sustainable Energy Reviews. 36, 220-227.
- 22.Balakrishnan C, Subha L, Neelakantan M A, Mariappan S S. (2015) Synthesis, spectroscopy, X-ray crystallography, DFT calculations, DNA binding and molecular docking of a propargyl arms containing Schiff base. , Spectrochimica Acta Part A 150, 671-681.
- 23.M J Frisch, G W Trucks, H B Schlegel, G E Scuseria, M A Robb et al. (2004) Gaussian Inc. , Wallingford, CT
- 24.H B Schlegel. (1982) Optimization of equilibrium geometries and transition structures. , J. Comput. Chem 3, 214-218.
- 25.M H Jamróz. (2006) Vibrational modes of 2,6-, 2,7-, and 2,3-diisopropylnaphthalene. , A DFT study, J. Mol. Struct 787, 172-183.
- 27.Michalska D, Wysokinski R. (2005) The prediction of Raman spectra of platinum(II) anticancer drugs by density functional theory. , Chem. Phys. Lett 403, 211-217.
- 28.Rauhut G, Pulay P. (1995) Transferable Scaling Factors for Density Functional Derived Vibrational Force Fields. , J. Phys. Chem 99, 3093.
- 29.Socrates G. (2001) Infrared and Raman Characteristic group Frequencies, Tables and Charts, third ed.,Wiley,Chichester.
- 30.John Xavier R, Dinesh P. (2013) Conformational stability, vibrational spectra, HOMO–LUMO and NBO analysis of 1,3,4-thiadiazolidine-2,5-dithione with experimental (FT-IR and FT-Raman) techniques and scaled quantum mechanical calculations. , Spectrochim. Acta A 113, 171-181.
- 31.Sumayya A, C Y Panicker, H T Varghese, Harikumar B. (2008) Vibrational spectroscopic studies and AB initio calculations of l-Glutamic acid 5-amide. , Rasayan J. Chem 1, 548.
- 32.Mushtaque M, Jahan M, Ali M, Khan M S, Khan M S et al. (2016) Synthesis, characterization, molecular docking, DNA binding, cytotoxicity and DFT studies of 1-(4-methoxyphenyl)-3-(pyridine-3-ylmethyl)thiourea. , J. Mol. Struct 1122, 164-174.
- 33.Socrates G. (2001) Infrared and Raman Characteristic Group Frequencies,third ed.,JohnWiley&SonsLtd.,Chichester.
- 34.Sebastian S, Sundaraganesan N, Manoharan S. (2009) Molecular structure, spectroscopic studies and first-order molecular hyperpolarizabilities of ferulic acid by density functional study. , Spectrochim. Acta A 74, 312.
- 35.Bellamy L J. (1959) . The Infrared Spectra of Complex Molecules,second ed.,John Wiley,New York,203 .
- 36.Varsanyi G. (1969) Vibrational Spectra of Seven Hundred benzene Derivatives. , Academic Press,New York
- 38.A E Reed, R B Weinstock, Weinhold F. (1985) Natural population analysis. , J. Chem. Phys 83, 735.
- 40.J P Foster, Wienhold F. (1980) Natural bond orbital analysis of near-Hartree–Fock water dimer. , J. Am. Chem. Soc 102, 7211-7218.
- 42.Alyar H, Kantarci Z, Bahat M, Kasap E. (2007) Investigation of torsional barriers and nonlinear optical (NLO) properties of phenyltriazines. , J. Mol. Struct 834, 516-520.
- 43.Castiglioni C, Del Zoppo M, Zuliani P. (1995) Experimental molecular hyperpolarizabilities from vibrational spectra in systems with large electron-phonon coupling. , Syn. Met 74, 171-177.
- 44.Zuliani P, Del Zoppo M, Castiglioni C, Zerbi G, Marder S R et al. (1995) Solvent effects on first‐order molecular hyperpolarizability: A study based on vibrational observables. Chem. Phys.103, 9935
- 45.Del Zoppo M, Castiglioni C, Zerbi G. (1995) A novel approach to estimate NLO response in organic conjugated molecules from vibrational spectra molecules with large values. , Non-Linear opt. 9 73.
- 46.Del Zoppo M, Castiglioni C, Zuliani P, Razelli A, Zerbi G et al. (1998) . , J. Appl. Polym. Sci 70, 73.
- 47.Ravikumar C, Huber Joe I, Jayakumar V S. (2008) Charge transfer interactions and nonlinear optical properties of push–pull chromophore benzaldehyde phenylhydrazone: A vibrational approach. , Chem. Phys. Lett 460, 552-558.
- 49.D R Kanis, M A Ratner, T J Marks. (1994) Design and construction of molecular assemblies with large second-order optical nonlinearities. Quantum chemical aspects. , Chem. Rev 94, 195-242.
- 50.T A Koopmans. (1934) Über die, Zuordnung Von, Wellenfunktionen and Eigenwerten zu, Elektronen Eines Atoms. , Physica 1, 104.
- 52.Kragh-Hansen U. (1981) Molecular aspects of ligand binding to serum albumin. , Pharmacol. Rev 33, 17-53.
- 53.G M Morris, Huey R, Lindstrom W, M F Sanner, R K Belew et al. (2009) Autodock4 and AutoDockTools4: automated docking with selective receptor flexibility. , Journal of Computational Chemistry 16, 2785-2791.
Cited by (10)
This article has been cited by 10 scholarly works according to:
Citing Articles:
Polycyclic Aromatic Compounds (2022) Crossref OpenAlex Semantic Scholar
Journal of the Indian Chemical Society (2022) OpenAlex Semantic Scholar
Polycyclic Aromatic Compounds (2021) Crossref OpenAlex Semantic Scholar
Molecular Diversity (2021) Crossref OpenAlex Semantic Scholar
Journal of Biomolecular Structure and Dynamics (2021) Crossref
G. Kirishnamaline, J. D. Magdaline, T. Chithambarathanu, D. Aruldhas, A. R. Anuf - Journal of Molecular Structure (2021) Semantic Scholar OpenAlex
Journal of Biomolecular Structure and Dynamics (2020) OpenAlex
Songül Eğlence-Bakır, S. Çelik, M. Şahin, A. Ozel, S. Akyuz et al. - Journal of Biomolecular Structure and Dynamics (2020) Semantic Scholar
Spectroscopy Letters (2019) OpenAlex
A. Eşme - Spectroscopy Letters (2019) Semantic Scholar Crossref