
High Performance Liquid Chromatographic-UV Method for Determination of Atorvastatin Calcium in Pharmaceutical Formulations
Abstract
The effectiveness of atorvastatin calcium in lowering cholesterol is dose-related. It is available in 10, 20, 40, and 80 mg film coated tablets. In order to ensure quality, safety and efficacy of tablets in formulations, the objective of this presented work was to develop a new high performance liquid chromatographic-UV method for quantitation of active atorvastatin calcium in pharmaceutical formulations. The method was based on reversed-phase high performance liquid chromatographic-UV separation of atorvastatin at detection wavelength of 246 nm using Acclaim 120 C18 reversed phase LC column (5 mm, 250×4.6 mm) with mobile phase of acetonitrile-dichloromethane-acetic acid (68.6: 30.6: 0.8 v/v/v) at a flow rate of 1.0 mL min-1 at 25°C. Different variables affecting chromatographic separation were carefully studied and optimized. The study results provided chromatogram of atorvastatin with retention time of 2.68 min. The calibration curve was linear over the concentration range of 15-300 mg mL-1. No interference was observed from common pharmaceutical excipients present in dosage forms. The proposed method was successfully applied to the determination of atorvastatin calcium in pharmaceutical formulations and proved to be significantly not different with reference method. The proposed can be used as an alternate method for routine quality control analysis of active atorvastatin in research, hospitals and pharmaceutical laboratories.
Article Information
- Received
- Accepted
- Published
Academic Editor: Ashish Kumar, Milan State University
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2017 Syed Najmul Hejaz Azmi , et al
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Corresponding author: Dr. Syed Najmul Hejaz Azmi, Higher College of Technology, Applied Sciences Department, Applied Chemistry Section, P. O. Box 74, Al-, Khuwair, 133, Muscat, Sultanate of Oman;, Tel.: —
Competing Interests
The authors have declared that no competing interests exist.
Funding
No specific funding statement was provided by the authors.
Data Availability
No data-availability statement was provided by the authors.
Acknowledgements
The authors are highly grateful to the Ministry of Manpower, Sultanate of Oman for providing necessary research facilities and to the Dean, Heads of the Applied Science Department and Chemistry Section, Higher College of Technology, Muscat for their continuous support and encouragement. The authors wish to express their gratitude to M/s National Pharmaceutical Industries Company, Oman for providing the gift sample of pure atorvastatin calcium.
Citation:
Introduction
Atorvastain calcium trihydrate (CAS No. 344423-98-9; M.W. 1209) is a white powdery substance chemically known as Calcium (3R,5R)-7-(2-(4-fluorophenyl)-5-(1-methylethyl)- phenyl-4-(phenylcarbamoyl)-3,5-dihydroxyheptanoate trihydrate 1. The drug is soluble in methanol having molecular formula of C66H68CaF2N4O10,3H2O.
Atorvastatin is an adjunct to diet for reduction of elevated total cholesterol. Atorvastatin selectively and competitively inhibits hepatic enzyme 3-hydroxyl- 3-methyl glutaryl-coenzyme A (HMGCoA) reductase, lowering plasma cholesterol levels by suppressing hepatic production of very low density lipoprotein and low density lipoprotein cholesterol. The effectiveness of atorvastatin in lowering cholesterol is dose-related. It is available in 10, 20, 40, and 80 mg film coated tablets. In order to ensure quality, safety and efficacy of tablets in formulations, it is important to develop new analytical method for quantitative analysis of drug in pharmaceutical formulations. Azmi and co-workers developed several analytical methods for the estimation and quality control analysis of cefixime 2 and citalopram HBr 3. With increasing regulatory strictness, the quality and safety of atorvastatin calcium is important. Owing to this reason, various analytical methods have been developed in pure and dosage forms such as spectrophotometry 4, high performance thin layer chromatography 5, 6, high performance liquid chromatography 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17 and liquid chromatography-mass spectrometry 18. Most of these analytical methods utilize expensive reagents 4, sophisticated instrumentations 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18 and more analysis time 5, 6. Published HPLC methods utilized higher column temperature 9, 12, 14, 15, 16, 17 and higher retention time 7, 8, 9, 10, 11, 13, 17. Higher temperature having the possibility of degrading the active drug and higher retention time will delay the analysis process. Therefore, it is required to develop a rapid and accurate high performance liquid chromatographic-UV method with lower retention time at ambient temperature. The main aim of the present work is to develop a new validated reversed phase high performance liquid chromatographic-UV method for the determination of atorvastatin calcium in pharmaceutical formulations. The method was validated as per International Conference on Harmonisation (USA) guidelines 19.
Experimental
Reagents and Materials
Solvents and reagents used were of high purity grade. Deionized water was obatined by double distillation and purification through milli-Q water purification system. Pure atorvastatin calcium reference drug was gifted by National Pharmaceutical Industries Company, Oman. 0.06% atorvastatin calcium was prepared in mobile phase of acetonitrile-dichloromethane-acetic acid (68.6: 30.6: 0.8 v/v/v) for proposed method. 0.02% atorvastatin calcium was dissolved in 0.005 M potassium dihydrogen phosphate-acetonitrile-methanol (39: 56: 5 v/v/v, pH 4.6) mobile phase for reference method. Both mobile phases were cleaned with nylon disc filter of 0.45 mm (Merck Millipore Ltd, Tullagreen, Ireland) and degassed by sonication before use for preparation of drug. Commercial tablets of Torvast 10 (National Pharmaceutical Industries Company, Oman) and Atorlip 10 (Cipla, India) were purchased from local market.
Hanna pH meter (USA) and Dionex-Ultimate 3000 high performance liquid chromatography (Thermo Scientific, Australia) were used for measuring pH and recording chromatogram.
High Performance Liquid Chromatographic Characteristics
Chromatography was performed in the isocratic mode on Dionex-Ultimate 3000 high performance liquid chromatography (Thermo Scientific, Australia) with a 20 µL sample injection loop (manual), HPG 3200 SD Pump, TCC 3000 SD column oven and WDM 3000 photodiode array UV-Visible detector. The output signal was monitored and integrated using Ver. 5.80 SR11 chromeleon Data System Software on an Acclaim 120 C18 reversed phase LC column (250 mm × 4.6 mm, i.d. 5 µm particle size).
Proposed Method for the Determination of Atorvastatin Calcium
A standard solution of atorvastatin calcium (0.6 mg mL-1) was prepared in a mobile phase of acetonitrile-dichloromethane-acetic acid (68.6: 30.6: 0.8 v/v/v). A HPLC column was cleaned and passed with said mobile phase. Into a series of 10 mL standard volumetric flask, different volumes (0.25-5.0 mL) of 0.06% atorvastatin calcium were pipetted and diluted up to the mark with said mobile phase. 20 μL of drug solution was injected into the sample port and mobile phase was pumped at a flow rate of 1.0 mL min-1 for a run of 5 min. The column temperature was maintained at 25̊C and the eluent was detected at 246 nm for HPLC separation of atorvastatin calcium. A calibration curve was constructed by plotting the corresponding peak area against the initial concentration of drug and the linear equation was obtained using Origin Pro6.1 software for the determination of active atorvastatin calcium in pharmaceutical formulations
Reference Method for the Determination of Atorvastatin Calcium
Different volumes (0.25-5.0 mL) of 0.02% atorvastatin calcium prepared in a mobile phase of 0.005 M potassium dihydrogen phosphate-acetonitrile-methanol (39: 56: 5 v/v/v, pH 4.6) were transferred into 10 mL standard volumetric flask and diluted up to the mark with the said mobile phase. HPLC column was treated with the respective mobile so that the base line became flat. The drug solution (20 μL) was injected into the sample port and mobile phase was pumped at a flow rate of 1.0 mL min-1 for a run time of 10 min. The column temperature was maintained at 25̊C and the eluent was detected at 246 nm for HPLC separation of atorvastatin calcium. The peak area was plotted against initial concentration of atorvastatin calcium for calibration graph. The linear regression equation was generated and used to estimate atorvastatin calcium in commercial dosage forms.
Assay of Atorvastatin Calcium in Commercial Dosage Forms by Proposed and Reference Methods
10 commercially available tablets of Torvast 10 and Atorlip 10 were weighed and uniformly powdered. The solid equivalent to 60 mg for proposed method (or 20 mg for reference method) of atorvastatin calcium was weighed and completely dissolved in 100 mL mobile phase of acetonitrile-dichloromethane-acetic acid 68.6: 30.6: 0.8 v/v/v (or 0.005 M potassium dihydrogen phosphate-acetonitrile-methanol 39: 56: 5 v/v/v for reference method). The mixture was filtered using Whatmann No. 42 filter paper (Whatmann International Limited, Kent, UK), cleaned with 0.45 mm millipore nylon disc filter (Merck Millipore Ltd, Tullagreen, Ireland) and sonicated. The tablet solutions were assayed subjected to the procedure for assay of atorvastatin calcium by proposed and reference methods. The amount of atorvastatin calcium was calculated using the respective linear regression equation.
Validation
The proposed method has been validated as per the International conference on Harmonization guidelines 19. Parameters such as linearity, limit of detection, limit of quantitation, precision, specificity, robustness, accuracy, and applicability were addressed for developing a new method.
Linearity was investigated at different concentrations of atorvastatin calcium. Each concentration level was tested 5 times and the peak area of the eluted drug was recorded. The linear regression equation and other statistical parameters were generated using OriginPro 6.1 Software.
 (1)
(1)
Limits of detection (LOD) and quantitation (LOQ) were calculated using the following expressions:
where S0 and b are standard deviation and slope of the calibration line, respectively.
The precision of the proposed method was tested at 2 concentration levels of 49.8 and 252.0 µg mL-1 atorvastatin calcium and independently analyzed 5 times repeatedly within a day (intra-day precision) and over 5 consecutive days (inter-day precision).
 (2)
(2)
The specificity of the proposed method was studied at 60 µg mL-1 atorvastatin calcium along with excipients such as glucose, fructose, lactose, sodium benzoate, starch, povidone, methyl cellulose and micro crystalline cellulose added in different volumes at selected % concentration. The peak area was recorded within ±2% at each addition of excipients. The maximum tolerance limit of excipients was calculated using the following equation:
Mass/Volume (g L-1 or mg mL-1) = Volume taken (mL) × % concentration (3)
The robustness of the proposed method was investigated at 60 µg mL-1 atorvastatin calcium with small variations in the optimized experimental values of pH, flow rate, temperature of the column and mobile-phase ratio. The peak area was recorded within ±2% at each deliberate change of chromatographic conditions.
The accuracy of the proposed method was tested by standard addition technique. One volume of the fixed concentration of tablet solution was spiked individually with different concentrations of the reference atorvastatin calcium solution in 10 mL standard volumetric flask and diluted up to the mark with respective mobile phase. Each level was analyzed repeatedly for 5 times for the determination of active atorvastatin calcium in tablet. The unknown concentration of atorvastatin calcium in tablet solution was determined by the calibration graph: either by extrapolation of the calibration line or by taking the ratio of the intercept to the slope.
 (4)
(4)
The accuracy of the proposed method was tested by direct method and compared with the reference method. The freshly prepared tablet solutions of Torvast and Atorlip containing atorvastatin calcium were independently analyzed 5 times at 60 µg mL-1 atorvastatin calcium by proposed and reference methods. The results of 2 methods (proposed and reference) were utilized to get experimental t and F-values at 95% confidence level and compared with tabulated t and F-values for significance of testing. If experimental t and F-values are less than tabulated t and F-values values 20, the proposed is likely to be not significantly different with reference method. The bias was calculated by interval hypothesis test based on % recovery values of 2 methods (proposed and reference). The bias is acceptable within ±2.0% of the recovery values 21, 22. The bias with (lower value) and (upper value) values were calculated by the following expression:
where and are mean recovery values at selected measurements, respectively. Sp is the pooled standard deviation and ttabis the tabulated one-sidedt-value at 95% confidence level.
Results and Discussion
The UV visible absorption spectrum of atorvastatin calcium solution in mobile phases of acetonitrile-dichloromethane-acetic acid (68.6: 30.6: 0.8 v/v/v) and 0.005 M potassium dihydrogen phosphate-acetonitrile-methanol (39: 56: 5 v/v/v; pH 4.6) were recorded in the wavelength range of 190-400 nm. The λmax of 246 nm was recorded in each mobile phase and thus used as UV detection wavelength in HPLC system for HPLC separation of atorvastatin calcium.
The solution stability of atorvastatin calcium in both mobile phases was checked by UV-visible spectrophotometry for a period of 1 day. The results showed an absorption peak at 240 nm with no change in the absorbance. The solution stability of the standard drug solutions was also tested using TLC plates coated with silica gel G (Merck Limited, Mumbai, India) and developed in acetonitrile: dichloromethane: acetic acid (45:20:0.5 v/v/v) and acetonitrile: 0.005 M KH2PO4: methanol (28.5: 5: 2.5 v/v/v) solvent systems. The results showed a single spot with Rfof 0.66 (acetonitrile: dichloromethane: acetic acid - 45:20:0.5 v/v/v) and 0.75 (acetonitrile: 0.005 M KH2PO4: methanol - 28.5: 5: 2.5 v/v/v) indicated that the drug solutions were stable for at least 1 day. Hence, the prepared drug can be analyzed within the stability time period of 1 day.
Method validation was performed on the best determined stationary phase i.e. C18 column (250 mm × 4.6 mm; i.d. 5 µm particle size). Separation with good resolution and low tailing factor (less than 2) were advantages of using C18 column.
The basic chromatographic conditions were optimized for developing a new HPLC-UV method for determination of atorvastatin calcium after testing different conditions of HPLC such as column temperature, components of mobile phase, proportion of mobile phase components, detection wavelength, and flow rate.
The mobile phase of acetonitrile-dichloromethane-acetic acid (68.6: 30.6: 0.8 v/v/v) was selected after preliminary trials using different compositions of mobile phase. The chromatogram obtained showed better eluted peak, less retention time and low tailing factor (Figure 1). It is clear from the figure that the retention time of atorvastatin calcium using the said mobile was 2.68 min. The low retention will fasten the analysis, hence selected the said mobile phase for HPLC separation of atorvastatin calcium.
Figure 1. Chromatogram of atorvastatin calcium (240 mg mL-1).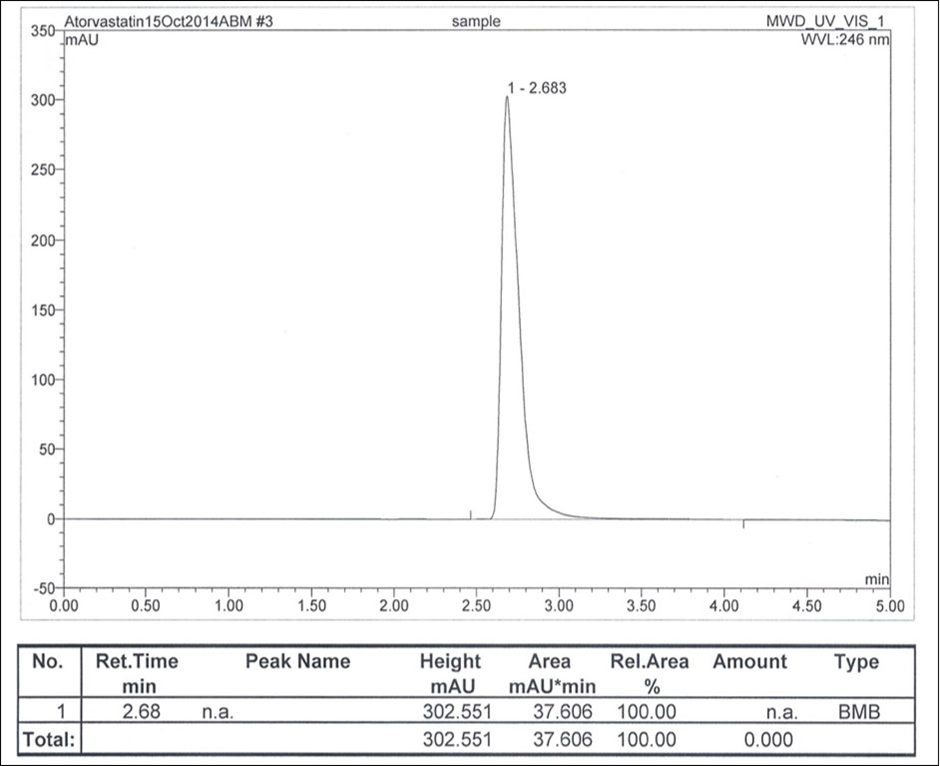
Download figure
Under theoptimized experimental conditions, peak areas at different concentrations of atorvastatin were obtained. The calibration data were inserted in the Origin Pro 6.1 software and utilized for obtaining linear regression equation with coefficient of correlation. The results are summarized in Table 1. It is evident from the table that the linear regression equation of A= 0.077 + 0.156 C was obtained with high coefficient of correlation (r = 0.9999). With this linear equation, the concentration of atorvastatin calcium was linear in the concentration range of 15-300 µg mL-1. Analytical parameters and chromatographic system suitability data of proposed and reference methods are given in Table 1. The results in the Table 1 supported the suitability of the proposed and reference methods. The experimental intercept of the proposed method was checked for significance of deviation from the theoretical intercept, zero using t = a / Sa 23 and found to be 0.726, which is less than the tabulated t-value (2.571,  =5) at 95% confidence level indicated an acceptable intercept. The acceptable intercept proved that the proposed method was procedurally error free.
=5) at 95% confidence level indicated an acceptable intercept. The acceptable intercept proved that the proposed method was procedurally error free.
| Reference method | Proposed method | Parameters |
|---|---|---|
| 246 | 246 | Maximum wavelength (nm) |
| 5-100 | 15-300 | Linear dynamic range (µg mL-1) |
| A= 0.101 + 0.669 C | A= 0.077 + 0.156 C | Linear regression equation |
| 0.08 | 0.106 | Standard deviation of intercept, Sa (µg mL-1) |
| 1.0×10-3 | 6.3×10-4 | Standard deviation of slope, Sb (µg mL-1) |
| 0.9999 | 0.9999 | Correlation coefficient (r) |
| 0.018 | 0.028 | Variance (S02) |
| 0.135 | 0.168 | Standard deviation of calibration line (S0) |
| 0.67 | 3.57 | Limit of detection (µg mL-1) |
| 1.96 | 10.82 | Limit of quantitation (µg mL-1) |
| 1.76 | 1.81 | Tailing factor |
| 6868 | 4418 | Theoretical plate |
| 7.57 | 2.68 | Retention time (min) |
Intra (within day) and inter day (between days) precisions were tested by determining the concentration of atorvastatin calcium at lower and upper concentration levels for 5 repeated times within the same day and on five consecutive days, respectively. The results are summarized in Table 2. It can be seen from the table that % relative standard deviation (RSD) values were in the range of 0.143-0.162 % for intraday and inter day precisions. The low % RSD values (intraday and interday precisions) at different concentrations showed that the proposed method was precise and can be used to analyze atorvastatin calcium in pharmaceutical formulations.
Table 2. Precision of the proposed method.The selectivity of the proposed method was investigated by testing the interference from common excipients such as glucose, fructose, lactose, sodium benzoate, starch, povidone, methyl cellulose and micro crystalline cellulose at 60 mg mL-1 of atorvastatin calcium. The peak area was recorded at varying concentrations of excipients and the results are summarized Table 3. Microcrystalline cellulose was tested and found to be insoluble in distilled water whereas methyl cellulose gave a viscous solution in distilled water. Therefore, both excipients were removed as added with drug in mobile phase and hence did not interfere in the determination process of atorvastatin calcium. The proposed method tolerated larger amount of excipients indicated that the proposed method was specific and selective, thus can be used to determine atorvastatin calcium in pharmaceutical formulations.
Table 3. Specificity and selectivity: Tolerated amount of excipients.| Excipients | Tolerance amount (mg mL -1) |
| Glucose | 3.50 |
| Fructose | 3.50 |
| Sodium benzoate | 1.44 |
| Lactose | 5.56 |
| Starch | 0. 12 |
| Povidone | 0. 12 |
The ruggedness of the proposed method was examined with a small change in the optimized data in the following manner.
volume of acetonitrile-dichloromethane-acetic acid (68.6: 30.6: 0.8 v/v/v, pH 4.5) (± 0.2 mL)
column temperature, 25 ˚C (± 1.0 ˚C)
flow rate, 1 mL min-1 (± 0.2 mL min-1)
In the above conditions, tablet solution of atorvastatin calcium (Torvast) at 60.0 µg mL-1 atorvastatin calcium was analyzed by the proposed method. The results showed mean % recovery of 100.04. Hence the proposed method was rugged and considered to be reliable for determination of active drug in tablet formulations.
The accuracy of the proposed method was tested by performing recovery experiments through standard addition technique. For this purpose, known portions of pure atorvastatin calcium were spiked with definite amount of Torvast solution (unknown) and the peak area was recorded. Standard addition graph was obtained by plotting the peak area versus concentration of added standard drug solution (Figure 2). The linearity of the regression line for Torvast tablet solution was good (r = 0.999). The intercept and slope of 9.414 and 0.157, respectively were obtained. The amount of atorvastatin calcium in Torvast tablet solution was calculated and found to be 59.86 µg mL-1 with % recovery of 99.76. The amount of drug in tablet solution was subjected to standard deviation, , and calculated using the following expression:
The value of was found to be 1.038 µg mL-1. The confidence limit for the concentration of atorvastatin calcium in Torvast was calculated by at n - 2 degrees of freedom and found to be 59.86 ± 1.038 µg mL-1. The most attractive feature of the proposed method by standard addition technique was its relative freedom from pharmaceutical additives and excipients and hence did not interfere with the determination process.
Figure 2. Standard addition plot: 1.0 mL of 0.06% atorvastatin calcium torvast tablet solution was spiked with 0, 0.1, 0.2, 0.3 and 0.4 mL standard solution of 0.06% atorvastatin calcium.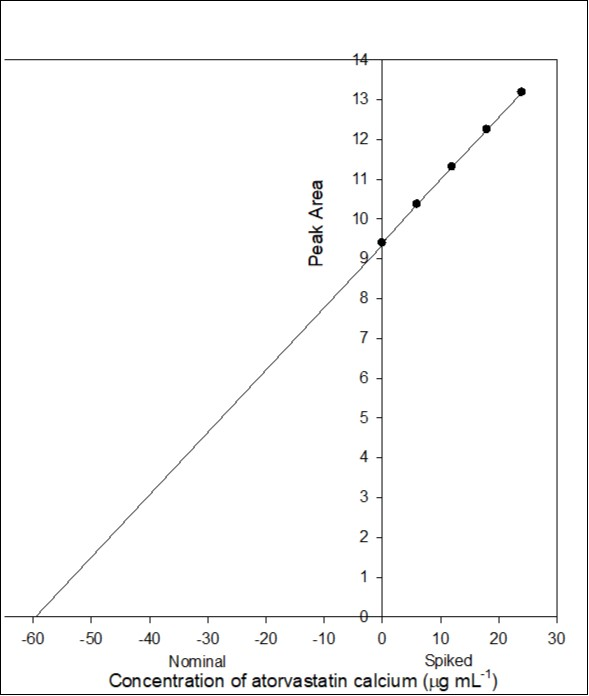
Download figure
The applicability of the proposed method for the determination of atorvastatin calcium in Torvast and Atorlip has been tested. The results of the proposed method were statistically compared with those of the reference method using point and interval hypothesis tests. t- and F-values at 95% confidence level were calculated using point hypothesis test and found to be in the range of 0.769-1.581 and 1.617-2.275, respectively. Both cases, t and F values were found to be less than the theoretical t and F values at 95% confidence level and hence proved that both methods were accurate and having no significant difference between them. The results are summarized in Table 4. Interval hypothesis test was utilized to calculate bias (lower limit, θL and upper limit, θU) in two methods and found to be in the range of 0.98-1.02 indicated the compliance of regulatory guidelines 21.
Table 4. Point and interval hypothesis tests: Applicability of the proposed method and significance of testing at 95% confidence level.The performance of the proposed HPLC method was compared with other existing HPLC methods (Table 5). As can be seen from the table, the linearity range was higher from the existing HPLC methods 7, 8, 9. Ambient temperature was used for preventing the possible degradation of active atorvastatin. Retention time was reasonably low as compared to the reported HPLC methods 7, 8, 9, 10 providing fast analysis of active atorvastatin in pharmaceutical preparations. The volume of acetonitrile in mobile phase was reasonable low as compared to other HPLC methods 8, 9. Hence, the overall performance of the proposed method was quite appreciable, and hence can be used as alternate method routine quality control analysis of active atorvastatin in commercial dosage forms.
Table 5. Comparison of the proposed HPLC method with other existing HPLC methods for the determination of atorvastatin calcium| Mobile phase | Temperature (⁰C) & UV detection wavelength (nm) | Linear range (μg mL-1) | Flow rate (mL min-1) & retention time (min) | References |
| Acetonitrile +dichloromethane + Acetic acid (68.6:30.6:0.8 v/v) | 25 & 246 | 15-300 | 1 & 2.68 | Proposed method |
| Ammonium dihydrogen phosphate, pH 5 (acidified with acetic acid) + methanol (60:40 v/v) | 25 & 240 | 2-12 | 1 & 6.91 | 7 |
| Potassium dihydrogen phosphate, 0.034 M (acidified with H3PO4 for pH 3.5) + Acetonitrile (30:70 v/v) | 25 & 254 | 10-50 | 1 & 4.8 | 8 |
| Potassium dihydrogen phosphate, 0.02 M (acidified with H3PO4 for pH 3.3) + Acetonitrile (30:70 v/v) | 30 & 280 | 5-30 | 1 & 3.2 | 9 |
| Ammonium acetate, 0.01M (pH 3) + Acetonitrile (50:50 v/v) | 25 & 254 | 4-400 | 1 & 19.3 | 10 |
| Potassium dihydrogen phosphate, 0.01 M (acidified with H3PO4 for pH 2) + Acetonitrile (72:28 v/v) | 25 & 247 | 20 to 200 | 0.6 & 1.92 | 11 |
| Sodium lauryl sulphate, 0.13 % (acidified with 0.06mL H3PO4) + Acetonitrile (72:28 v/v) | 55 & 210 | 20-160 | 1 & 2.15 | 12 |
| H3PO4, 0.1 % + Acetonitrile (55:45 v/v) | 25 & 230 | 3.2-12.8 | 0.35 & 2.89 | 13 |
| HClO4, 0.1 % (pH 2.5) + Acetonitrile (80:20 v/v) | 35 & 215 | 5-20 | 0.6 & 1.78 | 14 |
| Triethylamine buffer, 0.1 % (acidified with H3PO4 for pH 3) + Acetonitrile (40:60 v/v) | 45 & 240 | 56-104 | 0.8 & 1.62 | 15 |
| Ammonium acetate, 0.01M (pH 6.7) + Acetonitrile (42:58 v/v) | 40 & 245 | 50-150 | 0.2 & 0.68 | 16 |
| Potassium dihydrogen phosphate, 0.02 M + Acetonitrile + methanol (10:40:50 v/v) | 30 & 236 | 5-60 | 1.1 & 9.1 | 17 |
Conclusions
The proposed method was successfully applied for the determination of atorvastatin calcium in pharmaceutical formulations in the presence of excipients. The separation of the drug was achieved in 2.68 min. The proposed method was more specific, selective and rapid. It followed system suitability parameters such as tailing factor of 1.81 and theoretical plate of 4418. The ease of operation, sensitivity and reproducibility of the proposed method made it more suitable for routine quality control analysis of drug in research laboratories, hospitals and pharmaceutical industry.
References
- 2.Azmi SNH, Iqbal B, Al Mamari JK, Al Hattali KA, Al Hadhrami WN. (2014) Method development and validation for the determination of cefixime in pure and commercial dosage forms by spectrophotometry. , International Journal of Chemical, Nuclear, Metallurgical and Materials Engineering 6, 561-567.
- 3.Azmi S N H, Al-Fazari A, Al Badaei M, Mahrezi R. (2015) Utility of eosin Y as a complexing reagent for the determination of citalopram hydrobromide in commercial dosage forms by fluorescence spectrophotometry. , Luminescence 8, 1352-1359.
- 4.Ashour S. (2013) New kinetic spectrophotometric method for determination of atorvastatin in pure and pharmaceutical dosage forms. , Pharm. Anal. Acta 4, 232-238.
- 5.S J Varghese, T K Ravi. (2014) Quantitative simultaneous determination of fenofibrate, atorvastatin and ezetimibe in tablets using gradient high-performance column liquid chromatography and high-performance thin-layer chromatography. , J. Liq. Chromatogr. Relat. Tech 37, 2784-2799.
- 6.N M Rao. (2016) Development and validation of HPTLC method for the simultaneous estimation of amlodipine besylate and atorvastatin calcium in a combined dosage form. , Eurasian J Anal. Chem 11, 155-168.
- 7.Patil P M, Bobade A S. (2016) Development and validation of stability indicating RP-HPLC for determination of atorvastatin calcium and ezetimibe in bulk and pharmaceutical dosage forms. , Int. J. Pharm Pharmaceut. Sci 08, 38-42.
- 8.G K Swamy, Rao J V L N S, Kumar J M R. (2015) New stability indicating validated RP-HPLC method for simultaneous estimation of irbesartan and atorvastatin in combined tablet dosage forms. , Am. J. PharmTech Res 5, 380-389.
- 9.Khaleel N, S A Rahaman. (2016) Validated stability indicating RP-HPLC method for simultaneous determination of atorvastatin, fenofibrate and folic acid in bulk and pharmaceutical dosage form. , Der Pharmacia Lettre 8, 13-32.
- 10.Seshachalam U, C B Kothapally. (2008) HPLC analysis for simultaneous determination of atorvastatin and ezetimibe in pharmaceutical formulations. , J. Liq. Chromatogr. Relat. Tech 31, 714-721.
- 11.Vora D N, Kadav A A. (2008) Validated ultra HPLC method for the simultaneous determination of atorvastatin, aspirin and their degradation products in capsules. , J. Liq. Chromatogr. Relat. Tech 31, 2821-2837.
- 12.R K Seshadri, Desai M M, T V Raghavaraju, Krishnan D, D V Rao et al. (2010) Simultaneous quantitative determination of metoprolol, atorvastatin and ramipril in capsules by a validated stability-indicating RP-UPLC method. , Sci. Pharm 78, 821-834.
- 13.H O Kaila, M A Ambasan, A K Shah. (2011) A simple and rapid ultra-performance liquid chromatographic assay method for the simultaneous determination of aspirin, clopidogrel bisulphate and atorvastatin calcium in capsule dosage form. , Int. J. ChemTech. Res 3, 459-465.
- 14.Shetty S K, K V Surendranath. (2011) Stress degradation behavior of a polypill and development of stability indicating UHPLC method for the simultaneous estimation of aspirin, atorvastatin, ramipril and metoprolol succinate. , Am. J. Anal. Chem 2, 401-410.
- 15.Mallikarjuna S, Ramalingam P, Sriram P, Garima J, S K. (2013) Development and validation of stability indicating RP-UPLC method for simultaneous estimation of amlodipine besylate, and atorvastatin calcium in pharmaceutical dosage forms. , J. Liq. Chromatogr. Sep. Tech 4, 187.
- 16.Goel A, Baboota S, J K Sahni, K S Srinivas, R S Gupta et al. (2013) Development and validation of stability indicating assay method by UPLC for a fixed dose combination of atorvastatin and ezetimibe. , J. Chromatogr. Sc 51, 222-228.
- 17.Kumar P, Ghosh A, Chaudhary M. (2012) Stability indicating method development for simultaneous estimation of ezetimibe and atorvastatin in pharmaceutical formulations by RP-HPLC. , Pharma. Anal. Acta 3, 164-170.
- 18.Yacoub M, Abuawwad A, Alawi M, Arafat T. (2013) Simultaneous determination of amlodipine and atorvastatin with its metabolites, ortho and para hydroxy atorvastatin, in human plasma by LC–MS/MS. , J. Chromatogr.B,917-918: 36, 47.
- 19. (1995) International Conference on Harmonization ICH harmonised tripartite guideline- text on validation of analytical procedures. , Federal Register 60, 11260.
- 20.Mendham J, R C Denney, J D Barnes, Thomas M. (2002) Statistics: Introduction to Chemometrics. In: Vogel’s Textbook of Quantitative Chemical Analysis, 6th edition. Pearson Education. , Singapore, p: 137.
- 21.Hartmann C, Smeyers-Verbeke J, Pinninckx W, Y V Heyden, Vankeerberghen P et al. (1995) Reappraisal of hypothesis testing for method validation: detection of systematic error by comparing the means of two methods or of two laboratories. , Anal. Chem 67, 4491-4499.
Cited by (9)
This article has been cited by 9 scholarly works according to:
Citing Articles:
Journal of Liquid Chromatography & Related Technologies (2025) Crossref OpenAlex Semantic Scholar
Microchemical Journal (2024) OpenAlex Semantic Scholar
Sustainable Chemistry and Pharmacy (2023) OpenAlex Semantic Scholar
Journal of Chromatography Open (2022) OpenAlex Semantic Scholar
Trends in Sciences (2022) OpenAlex Semantic Scholar
Urvi Chotaliya, Hiteksha J. Dobariya, Disha L. Barad, A. Vyas, Dhruvanshi Gol - Analytical Chemistry Letters (2022) Semantic Scholar
M. R. Khan, M. Azam, A. Alammari - Journal of King Saud University - Science (2021) Semantic Scholar OpenAlex
International Journal of Applied Pharmaceutics (2019) OpenAlex Semantic Scholar
Journal of new developments in Chemistry (2017) OpenAlex