
Template Independent Synthesis of Nucleic Acid Libraries
Abstract
Though, directed evolution/In vitro evolution has greatly enhanced the applicability of natural biomolecules, there is still a big void in synthetic biology, which could be filled only when we are able to make novel/synthetic functional biomolecules. Terminal deoxyribonucleotidyl transferase (TdT) is the only known DNA polymerase, which can add deoxyribonucleotides without the requirement of a DNA template. Here, we are introducing the concept of Template-Independent Synthesis of Nucleic Acids (TISNA), where we have exploited the property of terminal deoxyribonucleotidyl transferase to add deoxyribonucleotides to the 3’ end of an oligonucleotide for the generation of de novo libraries of ssDNA, dsDNA coding sequences and RNA. We are able to generate libraries that have diversity not only in sequence but also in length in a single library itself. The length of double stranded random gene libraries generated using this approach ranges from 200 base pairs to 10 kilobase pairs. The ability to make random nucleic acid libraries from scratch (independent of any template information) in the laboratory could open up new avenues and holds promise for the pharmaceutical and biotechnological sectors.
Author Contributions
Academic Editor: Wentao Xu, Food safety and molecular biology
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2017 Kiran D. Bhilare, et al
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Introduction
There is no denying the fact that nucleic acids and proteins made after billion years of natural evolution are serving the host organism in the best way possible; still the applicability of the existing biomolecules for humans still has a lot to be desired 1. The first land mark work in the field of synthetic biology was the synthesis of the first complete gene, a yeast tRNA, as demonstrated by HarGobind Khorana 2. Few years ahead, synthesis of the first peptide- and protein-coding genes was done in the laboratories of Herbert Boyer and Alexander Markham, respectively 3, 4.
Szostak reported for the first time that genuinely new enzymatic activities could be created de novo without the need for prior mechanistic information by selection from a naive protein library of very high diversity 5, 6. The chemically synthesized library which they had generated was limited by the size as the length of randomized nucleotide addition was restricted only to 200-300 bases. To generate higher sized sequences, different parts (more than 150 bases) were made separately and later on attempted for ligation, but the overall yield was still marginal 7.
The biggest challenge in building novel nucleic acids de novo is the limitation of solid-phase DNA synthesis, which does not have the capacity to randomize beyond a couple of hundred bases 8. We envisaged that greener synthesis of novel random nucleic acid and protein libraries could be built in the laboratory only when nucleotide polymerization is done without relying on any specific template. Since the solid-phase DNA synthesis is able to generate random libraries of length not more than 200-300 bases, we looked for the enzymatic means to address this problem. Terminal deoxynucleotidyltransferase (TdT), due to its property of template-independent nucleotide addition, looked like the appropriate tool to build random libraries of nucleic acid molecules 9, 10. It has been reported in the literature that protruding, recessed or blunt-ended double or single-stranded DNA molecules serve as its substrate 11, 12. TdT, catalyzes the addition of dNTPs to the 3' endof single-stranded and double stranded DNA molecules 13, 14, 15, 16. Here, we present an approach to generate nucleic acids from scratch using terminal deoxynucleotidyltransferase (TdT) where we have successfully generated randomized single stranded DNA library, double stranded DNA library, RNA library and novel coding sequences.
Materials and Methods
Enzymes and Chemicals
The various biochemical such as TdT with 5x buffer, Pfu DNA polymerase with 10x Pfu buffer, Taq DNA polymerase supplied with 10x Taq buffer, magnesium chloride (MgCl2), dNTP Mix, dATP, dGTP, dCTP, and dTTP, fast digest NdeI, fast digest XhoI, 10x fast digest buffer, fast AP with 10X fast AP buffer, 6x Loading dye, 1 kbp ladder, nuclease free water were purchased from Fermentas (Thermo Fischer Scientific, Waltham, U.S.A.). Agarose was procured from Invitrogen Life Technologies (Thermo Fischer Scientific, Waltham, U.S.A.). All other chemicals used were of molecular biology grade.
Primers
Both the primers were purchased from Integrated DNA Technologies (IDT), Iowa, U.S.A. We envisaged that synthetic gene libraries should start with a start codon and end with stop codons flanked by restriction sites on both the 5’ and 3’ ends to assist in cloning. The initiation oligonucleotide (F1) was designed in such a way that it had ATG (start codon) on the 3’ end and a restriction site upstream of the start codon. Sequence of the starting oligonucleotide was 5’-CGACCGAAATACCTGCTGCCATATG-3’; where ATG is the start codon and CATATG is the restriction site for the NdeI enzyme. The reverse oligo-dT primer (R1) was designed in such a way that it had stop codon in three frames and restriction site is kept upstream of the stop codons. The sequence of the XhoI-oligodT reverse primer (R1) was 5’-AGGGCCTCGAGTCATTCATTCA(T)18-3’, where CTCGAG is restriction site of XhoI and TCATTCATTCA incorporates stop codons in three reading frames after annealing to the adenylated single stranded DNA during the primer extension step.
Kits
Plasmid Miniprep kit, Gel extraction kit, PCR purification kit/ Enzymatic clean up kit were purchased from Fermentas (Thermo Fischer Scientific, Waltham, U.S.A.) and Invitrogen Life Technologies (Thermo Fischer Scientific, Waltham, U.S.A.). pENTRTM/TEV/D-TOPO Directional Cloning Kit with One ShotR Mach1TM-T1R chemically competent E. coli cells was ordered from Invitrogen Life Technologies (Thermo Fischer Scientific, Waltham, U.S.A.).
Generation of Randomized Nucleic Acid Library
The final reaction mixture for single strand synthesis roughly consisted of OligoTdT-NdeI primer, deoxynucleotides, 1X terminal transferase buffer and terminal transferase enzyme (30 U). The reaction mixture was incubated at 37 °C for different time intervals and stopped by heating the mixture at 70 °C for 15 min. Enzymatic clean up kit was used for the purification of single stranded DNA (ssDNA) library. The purified single stranded DNA library was adenylated using terminal deoxynucleotidyl transferase, followed by the primer extension step using (R1) reverse primer. Around 1 μg purified single stranded DNA was incubated at 25 °C with 30 U of TdT in the presence of 1 mM dATP and 1X TdT buffer 0.1 M potassium cacodylate 12.5 mM Tris 0.5 mM CoCl2 0.005% Triton X. The reaction was incubated at 37 ºC for 5 min and then heat-inactivated at 70 ºC for 15 min. The primer extension reaction was combined with the polymerase chain reaction (PCR) by adding both the forward primer (F1) and reverse primer (R1) in the PCR reaction mixture as shown in Figure 1. The final concentration of forward as well as reverse primer was 0.5 μM. PCR was carried out using 100 ng of single stranded polyadenylated library with 2 U Pfu DNA polymerase (Fermentas), 200 μM of dNTP , 1X Pfu buffer (20 mM Tris-HCl, 10 mM (NH4)2SO4, 10 mM KCl, 0.1% Triton X-100, 2 mM MgSO4, 0.1 mg/mL BSA, pH 8.8). Thermal program was set at 94 °C for 5 min (initial denaturation), followed by 30 cycles of denaturation at 94 °C for 1 min, annealing at 56 °C for 1 min, extension at 72 °C for 1.5 min and final extension at 72 °C for 10 min. The final product was purified using gel extraction kit. It was eluted in nuclease free water and stored at -20 °C for future use.
Figure 1.Schematic representation of Template Independent Synthesis of Nucleic Acids (TISNA).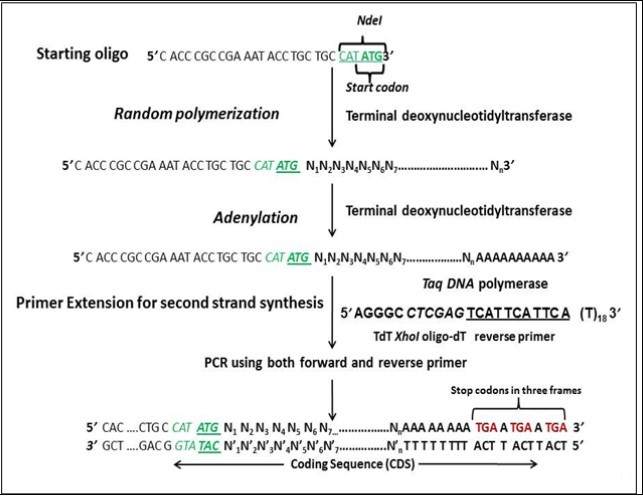
Generation of RNA Library through TISNA
The proposed scheme for the synthesis of double stranded DNA library was also pursued for the synthesis of another type of nucleic acid material, i.e. RNA. For generating random RNA libraries, a 25 base primer was designed with T7 promoter sequence 5’-TAATACGACTCACTATAGGG-3’ and used terminal deoxynucleotidyl transferase to generate single stranded DNA library on it. This single stranded DNA library was subsequently made into a double stranded DNA (ds DNA) library and used as a template for in vitro transcription (Transcript Aid T7 High Yield Transcription Kit, Thermo Fischer Scientific, Waltham, U.S.A.). Double stranded linear DNA is a prerequisite for any in vitro transcription reaction. Approximately 1 μg DNA library was incubated with 2 μL Transcipt Aid Enzyme mix, 10 μM each of rNTPs (ATP/GTP/CTP/UTP), 1X Transcript aid reaction buffer in 20 μL reaction mixture. Samples were incubated at 37 °C for 6 h. RNase A (final concentration 50 µg/mL) treated samples were further incubated at 37 °C for 1 h.
TOPO Cloning
The double stranded library (coding sequences) thus generated in the above steps was cloned following the manufacturer’s protocol as mentioned in the pENTR Directional TOPO Cloning Kit. The final ligation mixture contained 1 μL pENTR/TEV/DTOPO vector, salt solution (final concentration 0.2 M NaCl, 0.01 M MgCl2) with 50 ng dsDNA library. The ligation mixture was incubated at 23 °C for 10 min.
Transformation of One Shot E. coli MACH1TMT1R Chemically Competent Cells
The above ligation mixture was added to the vial of E. coli MACH1TM-T1R chemically competent cells. It was incubated on ice for 10 min. The cells were the subjected to heat shock at 42 °C for 30 seconds followed by the addition of 250 μL SOC medium. The tube was tightly capped and kept for shaking at 37 °C, 200 rpm for 1 h. The transformed cells were plated on Luria Bertani Broth (Miller) containing 1.5% (w/v) agar Kanamycin plates. The clones thus obtained after 12 to 14 h were grown in Luria Bertani Broth (Miller) in the presence of Kanamycin (35 μg/mL) as the selection marker.
Sequencing and Alignment Search of the Open Reading Frames (ORFs)
Positive clones were sequenced at Scigenome, Bangalore, India. Multiple alignments of clones was performed using CLUSTALW software.
Results
Generation of Template Independent Single as well as Double Stranded Nucleic Acid Random Libraries
Encouraged by the template independent polymerase activity of TdT, synthesis of random library of single stranded DNA molecules was attempted using a random hexamer as the starting molecule. During which, it was found that in the presence of dNTPs, TdT was capable of adding nucleotides to the random hexamer creating a random library of single stranded DNA molecules. Also, the process was affected by incubation time and primer:dNTP ratio. When we succeeded in our first attempt, our next goal now was to make double stranded DNA molecules. For this, phi29 DNA polymerase was used to catalyze primer extension on single stranded library to create a random library of double stranded DNA molecules but it gave non-reproducible and erratic results during every reaction. On the contrary, terminal transferase proved to be the suitable enzyme for single strand DNA synthesis as well as for adenylation reaction. .. The initiation oligonucleotide was designed in such a way that it had a start codon at the 3’ end and a restriction site upstream of it (Figure 1). F1 oligonucleotide was designed with NdeI site since its recognition sequence contains a start codon at its 3’ end. The reverse oligo-dT primer (R1) incorporates stop codons in three reading frames after annealing to the adenylated single stranded DNA library. This resulted in the generation of random libraries of coding sequences as there is no control on the course of polymerization reaction. Also, increase in the ratio of initiation oligonucleotide pmols to pmols of dNTP resulted in a decrease in the length of ssDNA library (Figure 2). When 2 pmols oligonucleotide (Figure 2, Lane 3) was used the gene library developed was from 2 kbp to more than 10 kbp (In fact, the DNA is so large in size that it hasn’t come out of the well). When 5 pmols oligonucleotide (Lane 4) was used the gene library developed was from 1 kbp to more than 10 kbp. When 10 pmols oligonucleotide (Lane 5) was used the gene library developed was from 500 bp to 10 kbp. When 100 pmols oligonucleotide (Lane 6) was used the gene library developed was from 100 bp to 1 kbp. The reason behind this observation is that the concentration of dNTPs is kept the same in all the examples and only the amount of oligonucleotide has been varied. As a result, we are getting the ratio of pmols ends to dNTP concentration as 1/100 for 100 pmols oligo concentration, 1/1000 for 10 pmols, 1/2000 for 5 pmols concentration and 1/5000 pmols fo 2 pmols concentration. This roughly means that for 100 pmolss, for 1 molecule of oligonucleotide, 100 molecules of dNTPs can be added by terminal transferase enzyme. Hence we see lengths in increasing order, i.e: 1/5000 > 1/2000 > 1/1000 > 1/100.
Figure 2.Generation of random libraries of ssDNA. TISNA approach was used to generate library of ssDNA molecules. Lane 1 100 bp ladder, Lane 2 Used F1 oligo, Lane 3-6 reaction containing ratios of 1:5000 , 1:2000, 1:1000 and 1:100 respectively, Lane 7 1 kbp ladder
The length of single stranded DNA library progressively increased with the increased reaction time (Figure 3). In case of 3h reaction time, relatively shorter length libraries are formed as compared to 6h, 12h and 24h time reactions. Beyond 6h incubation time, the reaction reaches saturation and hence, there is not much increment in the length of the nucleic acid library as observed in the gel image.
In the gel image, the gel purified double stranded DNA library appeared as a smear having size ranging from 250 base pairs upto 10.0 kilobase pairs (Figure 4).
Figure 3.Optimization of library size with respect to time of incubation. Lane 1 initiation oligo F1, Lane 2 100 bp ladder, Lane 3-6 Reactions were incubated for 3, 6, 12 and 24 h respectively, Lane 7 1 kbp ladder
Figure 4.Generation of random library of dsDNA from ssDNA library. Lane 1 1 kbp ladder, Lane 2 PCR reaction run after the primer extension step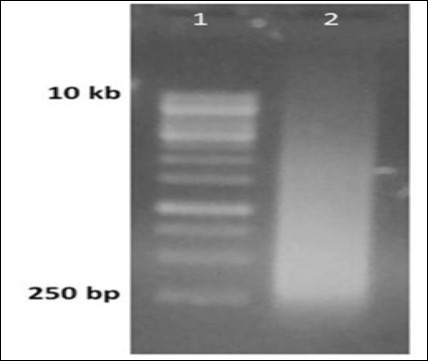
Generation of Template Independent RNA Library
After generating in vitro dsDNA coding sequences, we used it for generating random libraries of RNA. In this case, we used the F1 oligonucleotide having a T7 promoter sequence and terminal deoxynucleotidyl transferase to generate ssDNA library on it. After running on 1 % (w/v) agarose gel, random RNA libraries were visible (Figure 5). The RNA from the sample was verified since after RNase A digestion, there was no RNA band in the treated sample.
Figure 5.Generation of RNA library through TISNA. Lane 1 dsDNA template used in the reaction, Lane 2 1 kbp ladder, Lane 3 In vitro transcription kit reaction, Lane 4 Rnase A treated In vitro transcription reaction
Sequence Analysis of Random Coding Sequence Library Members
The careful analysis of the sequence for the clones obtained using Template Independent Synthesis of Nucleic Acid (TISNA) revealed that guanine and cytosine are uniformly present (% GC is ~50%) in all the synthetic DNA molecules. The statistical analysis (% individual and % dinucleotide pair: AA, AC, AG, AT, CC, CG, CT, CA, GA, GG, GC, GT, TA, TT, TC, TG) for the present clones has been done and reported in the “Supplementary Table 1”. The multiple alignment of the clones revealed high degree of diversity among the different cloned synthetic DNA molecules (Figure 6). There exists no dT in the analyzed sequence because we have used the double stranded library that was made using AGC combination only. This was done deliberately to obviate the incorporation of any stop codon (namely-TGA, TAG, TAA) in the sequence of “open reading frame, ORF” that we intend to generate using TISNA approach.
Figure 6.Coding sequences generated through Template Independent Synthesis of Nucleic Acids (TISNA). The sequence of the reverse primer is underlined while the stop codons in three frames are highlighted in red.
Discussion
Gene synthesis is a provocative field having multiple implications in synthetic biology as well as in molecular biology. The assembly of DNA sequences from oligos finds applications in DNA synthesis, gene expression and in vitro mutagenesis 17. The development of new antibiotics by de novo-designed polyketide synthase gene clusters was reported in 2005 18. It has also been reported that the DNA polymerases of thermophilic bacterium Thermus thermophilus and Thermococcus litoralis was used to synthesize long stretches of linear double stranded DNA (upto 200 kbp) de novo in the complete absence of primer and template DNA 19. The synthesized DNA fragments were tandem repetitive sequences and none of them was a coding sequence. Those random DNA It fragments were attempted for cloning although they got success to clone inserts of only smaller size. This group demonstrated that DNA sequences can be made abinitio by protein even in the absence of any pre-existing genetic information but the process was a slower one. In another work, de novo synthesis of DNA was observed in the presence of restriction endonuclease-EcoRI under relatively high temperature, the process termed as cut-grow DNA synthesis 20. They have mentioned that, EcoRI was used to digest double-stranded DNA, which was later amplified in the presence of DNA polymerase and dNTPs. Template-free and primer independent DNA upto several hundred kb was synthesized in the presence of DnaB, a bacterial DNA polymerases I obtained from E.coli 21. The % GC content in the final product was far more than % AT content. Our results differ from this group that in the present case we have an unbiased proportion of GC over AT content (as mentioned in the supporting information) and we have created open reading frames that too from a short primer without the inclusion of external template DNA at any step. In an European Patent, a method for producing gene libraries is disclosed wherein DNA strands are inserted into the genes to be mutated, in various positions 22. It involved the incorporation of a transposon (Donor) into said DNA (Gene) at different random positions. In vivo method for the construction of randomized gene libraries by homologous recombination using the gamma subunit of Kluyveromyces lactis killer toxin, was mentioned in another European patent 23. Both the above methods suffer from the limitation that they created variants by randomizing a specific region in that gene. The libraries prepared in EP1385949B1 and EP1613751A1 were merely variants of the same gene. Thus in the literature survey, there exist no approach to synthesize all the three forms of nucleic acids and coding sequences (random libraries) from scratch. The generation of synthetic genes whose protein products could catalyze a wide range of different reactions, is a long-standing goal of researchers in the field of synthetic biology 24. Generating new genes and proteins would require a strategy where we are able to generate diverse libraries of nucleic acids from scratch. There is a room for the development of technology that allows for the creation of novel DNA molecules in a single step without resorting to any pre-existing DNA. TISNA approach uses an initiation primer as a starting point for random nucleotide polymerization, which is catalyzed by terminal deoxynucleotidyl transferase (Figure 1). The enzyme terminal deoxynucleotidyltransferase catalyzed the nucleotide addition on an initiation oligonucleotide having ATG (start codon) at the 3’ end. TdT could build coding sequence libraries of different sizes ranging from 100 to 10,000 bp. Although in the present case, oligodT based approach has been used for converting our single stranded DNA library into double stranded DNA library, researchers can use other strategies to get a primer binding sequence on the random 3’ ends. This will certainly avoid the unwanted stretches of adenine before stop codons at the 3’ end of the library. While making coding sequences, several combinations of all the four deoxyribonucleotides were tried (Figure 7).
Nucleic acid libraries were made in the absence of either dTTP or dATP while keeping the rest of the three deoxyribonucleotides in equimolar concentrations. Another reaction was also tried with 50 μM dATP, 50 μM dTTP, 200 μM dCTP and 200 μM dGTP for TdT mediated template independent polymerization (Figure 7). In Figure 4 the size of most dsDNA is above 250bp, but in supplementary table 1 the length of all the clones are smaller than 250bp. During the ligation between the linearized vector and the entire library (comprising of dsDNA of all size range), smaller sized ORFs must have outcompeted large size products. In order to get rid of this, efforts are in place to synthesize only larger sized ORFs (more than 750bp).
Figure 7.Generation of random dsDNA library with various combinations of nucleotides. a) Lane 1 ssDNA template used in the reaction, Lane 2 F1 initiating oligo, b) Lane 3 1kbp ladder, Lane 4-6 ds DNA library made using TGC, A25T25G100C100, ATGC combination respectively
Conclusions
Terminal transferase is a pretty robust enzyme that is primarily used for end labeling and addition of homopolymer tails 25. However, it was not until the demonstration of its role in certain types of leukemic lymphocytes that interested its usage in the molecular biology 26. This is the first instance where tTdT has been utilized for the generation of huge libraries of single stranded DNA, double stranded DNA/coding sequences and RNA library. The naturally occurring dNTPs that are used in the present case can be replaced by other modified or unnatural nucleotides to change the works perspective in an entirely different direction. Nucleic acid libraries generated using TISNA based approach have huge length and sequence diversity (Figure 1). The single stranded DNA and RNA libraries can be exploited to screen for deoxyribozymes and ribozyme like activity, respectively. The increase in length may provide the single stranded DNA and RNA much more structural complexity strengthening their suitability as catalysts. Hitherto, with the help of TISNA approach, novel coding sequence libraries could be generated. The ability to generate more such novel coding regions will help researchers to untap the protein or sequence spacestill elusive to us.
Compliance with Ethical Standards
Ethics approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Supporting information Percentage GC content analysis of the ORFs of cloned library members obtained in this study.
Supplementary Data
Acknowledgements
We are highly thankful to Swapnil Sinha for her kind help regarding the multiple sequence alignment as well as in the analysis of gene sequences (ORFs) and Mahesh Patil for his thoughtful corrections in the present manuscript. Kiran Bhilare is a recipient of Senior Research fellowship from the Department of Biotechnology, Government of India.
References
- 2.H G Khorana, K L Agarwal, Bagchi H, M H Caruthers, N K Gupta et al. (1972) . , J. Mol. Biol 72, 209-217.
- 3.M D Edge, A R Greene, G R Heathcliffe, P A Meacock, Schuch W et al. (1981) . , Nature 292, 756-762.
- 10.Coleman M S, Hutton J J, P De Simone, Bollum F J. (1974) . , Proc. Natl. Acad. Sci. USA 71, 4404-4408.
- 11.E A Motea, A J Berdis. (2010) . , Biochim. Biophys. Acta. (BBA)-Proteins and Proteomics 1804, 1151-1166.
- 18.H G Menzella, Reid R, J R Carney, S, S J Reisinger et al. (2005) . , Nature Biotechnol 23, 1171-1176.
Cited by (2)
This article has been cited by 2 scholarly works according to:
Citing Articles:
ChemBioChem (2023) Crossref
ChemBioChem (2022) OpenAlex