
Functional, Structural and Contextual Analysis of a Variant of Uncertain Clinical Significance in BRCA1: c.5434C->G (p. Pro1812Ala)
Abstract
Interpreting variants of uncertain significance (VUS) for their effect on protein function, and therefore for the risk of developing cancer, has become a challenge in clinical practice for genetic counselling services. The present work combines structural bioinformatics and systems biology based mathematical modelling approaches with the aim of determining the pathogenicity of the mutation c.5434C->G (p.Pro1812Ala) in the BRCA1 gene (detected in a patient from a high risk family) and also to mechanistically understand the effect of this mutation in DNA damage response, a key process in cancer development. The results obtained showed that this mutation prevents the interaction of BRCA1 with key proteins of the cell cycle, subsequently impairing BRCA1-dependent induction of cell cycle arrest. The comparison of the molecular mechanisms associated with the native BRCA1 protein and the mutated variant function in DNA damage response showed that the latter undergoes a reduction in its ability to modulate pathways that are critical for DNA repair and cell cycle control. Therefore, this variant will not be able to exert its tumor suppressive action. Interestingly, these conclusions can be extrapolated to all mutations that, like c.5434C>G (p.Pro1812Ala) BRCA1, cause loss of BRCT domain activity.
Author Contributions
Academic Editor: Xi Zhang, Co-founder & Scientist SinoScript LL
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2017 Rafael Morales, et al.
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Introduction
Deleterious variants in the BRCA1 and BRCA2 genes account for approximately 20% of cases of hereditary breast and ovarian cancer. The BRCA1 gene is found on the long arm of chromosome 17 (17q21) and plays a crucial role in DNA damage response. BRCA1 inactivating mutations lead to genetic instability, indirectly causing tumours to occur as a result of an accumulation of mutations in cell cycle regulatory genes. In Spain, families that carry the deleterious mutation in the BRCA1 gene have a 52% cumulative risk of developing breast cancer and a 22% risk of developing ovarian cancer by the age of 70 1.
The BRCA1 protein is a protein of 1863 amino acids. The two most important domains of BRCA1 are the RING domain and the BRCT domain. The RING domain is located at the amino terminus of BRCA1, between amino acids 1 and 109 (exons 2 - 7), and it is responsible for the E3·ubiquitin·ligase activity of BRCA1 2 and binds to the BARD1 protein. The BRCT domain mediates binding with phosphorylated proteins such as: Abraxas 3, 4, BACH1/BRIP1 5, and CtlP 6, forming three mutually exclusive protein complexes named A, B and C complexes (see below). A number of other proteins also bind to the C-terminal region of BRCA1 and/or its central region 7 (Figure 1).
Figure 1.BRCA1 domains and interacting proteins: BRCA1 contains a RING domain at its N-terminus, two BRCT domains at the C-terminus and a coiled-coil domain upstream of BRCT domains. The interacting proteins are shown under the region of BRCA1 required for their association.
BRCA1 is involved in several steps of the DNA damage response process, a cell cycle control mechanisms that prevent the progression of the cell cycle while activating DNA repair routes. From a mechanistic point of view, it is known that BRCA1 forms part of 4 macro-complexes 8, 9 BRCA1A, BRCA1B BRCA1C and BRCC. The BRCA1A complex acts as a DNA damage sensor and regulates the G2/M cell cycle checkpoint. The BRCA1B complex participates in the G1/S and intra-S-phase checkpoints, ensuring genome integrity prior to DNA synthesis. The BRCA1C complex mediates DNA repair through homologous recombination, facilitating the formation of single-stranded DNA in the step prior to homologous recombination. The BRCC complex is capable of recognising the previously formed single-strand DNA and mediating strand invasion; it is considered the effector in homologous recombination.
Interpreting variants of uncertain significance for the effect of protein function, and therefore for the risk of developing cancer, has become a challenge in clinical practice for genetic counselling services. It is necessary to develop tools that will help us reduce the high percentage of variants of uncertain significance, therefore facilitating genetic counselling process.
The variant of uncertain significance c.5434C->G (p.Pro1812Ala = Pro1812Ala) has been found in a high-risk family, where the proband is a woman diagnosed with papillary serous cystadenocarcinoma of the ovary, and her mother and maternal aunt are diagnosed with breast cancer. The c.5434C>G variant is found at position 28 of exon 23 of the BRCA1 gene. In this work we apply systems biology based mathematical modelling approaches to mechanistically determine the pathogenic effect of this variant.
Methods
The assessment of the physiological consequences of the c.5434C->G (p.Pro1812Ala) mutation with regard to its predisposition to develop cancer was approached by two complementary computational perspectives: a structural approach and a systems biology based functional approach.
Structural Bioinformatics Analysis
A three-dimensional structure of the BRCA1 protein was modelled after experimentally obtained structures contained in PDB, taking into account the changes induced by the mutation according to published literature 10, 11. Then, a molecular dynamics simulation in a system of interacting particles was run to simulate the variability of the BRCA1 in the cytoplasm and predict its time evolution. The resulting structures with the lowest values of free energy were selected, under the assumption that they are more stable and, hence, may be found in vivo. This exercise was conducted using CHARMM22 12 force field and calculating the partial charges through the Gasteiger method.
The presence of significant structural changes in the BRCA1 protein was detected through statistical parameters and subjective assessment after visualization and the stability of the mutated protein was determined based on certain key values, including potential energy, RMSD, and Ramachandran outliers. The predictions were compared with the conclusions of published studies.
Systems Biology Based Functional Approach
Therapeutic Performance Mapping System (TPMS) technology (Anaxomics Biotech, Barcelona) 13 was applied to generate and analyze mathematical models able to simulate in silico DNA damage response in the context of a native (wt) BRCA1 or the studied variant. Briefly, DNA damage response was first characterized at a molecular level through hand curated bibliography search. Then, a protein interaction map based on the human protein-protein interaction network was generated. The map was extended by adding knowledge-oriented connectivity layers, i.e., protein-to-protein interactions, including physical interactions and modulations, signalling, metabolic relationships, and gene expression regulation. Data was obtained from public and private external databases (Binding Database 14, BioGRID 15, IntAct 16, REACTOME 17, 18 and the manual curation of scientific literature. In the case of the mutated BRCA1 model the impaired protein interactions due to the mutation were restricted when constructing the protein network.
This static map was transformed into a dynamic mathematical model with predictive capacity through training them with functional information collected in Anaxomics proprietary Truth Table database. In brief, to train the mathematical models a collection of known input-output physiological signals was used. The molecular description of these input-output physiological signals mainly derives from literature mining and a compendium of databases that accumulate biological and clinical knowledge, such as microarray databases (e.g. GEO 19, 20) and drug databases (e.g. DrugBank 21). The methods applied rely within the field of Artificial Intelligence, including artificial intelligence graph theory and statistical pattern recognition techniques; genetic algorithms, artificial neural networks, dimensionality reduction techniques; and stochastic methods like Simulated Annealing, Monte Carlo among others 22.
Once the models had been generated, they were analysed in two sequential steps: First the mechanism of action of functional protein and the mutated protein in DNA damage response was evaluated independently, as detailed in the literature 23, and secondly the two individual mechanisms of action obtained were compared in order to assess the impact of the mutation on DNA damage response activity.
Results
In order to determine the physiological consequences of BRCA1 c.5434C->G (p.Pro1812Ala) mutation, this variant was analysed from two complementary perspectives to determine, first, the protein structural changes caused by the mutation in BRCA1 and then, the impact of these changes in the protein functionally in the context of the DNA damage response.
Structural Analysis of BRCA1 c.5434C->G
It has been previously reported that the c.5434C->G (p.Pro1812Ala) mutation causes an important splicing defect in BRCA1, resulting in the omission of exon 23, which in turn leads to the appearance of a premature stop codon (p.Gly1803GlnfsX11) 10. The resulting protein is truncated and lacks the second BRCT domain. The mutated protein BRCA1 is therefore shorter than the wild-type protein due to interruption of the BRCT domain. However, according to the results obtained in our structural analysis, comparison of the wild-type protein and the mutated protein shows that both variants of the protein seem to have the same secondary and tertiary structure. Deletion of the second BRCT domain does not appear to bring about changes in the secondary and tertiary structures of the rest of the protein (Figure 2).
Figure 2.Comparison between the 3D protein structure of functional BRCA1 (in green) and mutated BRCA1 models (in blue)
Both proteins have the same number of α helices, β strands and turns 24. The spatial distribution of the secondary structure is essentially the same, with slight differences that could be attributed to the molecular dynamics simulation. Domains other than BRCT, such as RING, NLS and Coiled Coil domains, do not seem to be altered.
Additionally, in order to evaluate whether the structural similarity between the variants remains invariable over time (i.e. if they are equally stable), we tracked the conformational changes through molecular dynamics during 100 picoseconds (Figure 3). The simulation confirmed that the variant remains stable through time. The values of free energy remained low along the simulation and the RMSD, which measures the similarity in the 3D structure, did not vary. That is, despite the disruption of the second BRCT domain, the rest of the protein (including RING, NLS and coiled coil domains) remains functional.
Figure 3.Dynamic Protein Model of the BRCA1 Pro1812Ala Variant
Mechanistic Comparison Between Wild-Type and Mutated BRCA1
Mathematical models simulating DNA damage response related mechanisms were used to compare two molecular scenarios: expression of the wild type and the c.5434C->G (Pro1812Ala) variant. Working on the basis that loss of the second BRCT domain disturbs the whole BRCT region 25, all interactions associated with this domain were removed from the protein-protein interaction network. The full list of protein-protein interactions that have been deleted can be found in Supplemental data 1.
Supplemental data 1. Restrictions used in the construction of the mutated BRCA1 model| Uniprot A | Gene A | Restriction | Uniprot B | |
| P35869 | AHR | does not activate | P38398 | BRCA1 |
| P38398 | BRCA1 | does not activate | Q92793 | CREBBP |
| P38398 | BRCA1 | does not activate | Q96RL1 | UIMC1 |
| P38398 | BRCA1 | does not activate | P15311 | EZR |
| P78347 | GTF21 | does not activate | P38398 | BRCA1 |
| Q9NWV8 | BABAM1 | does not activate | P38398 | BRCA1 |
| P38398 | BRCA1 | does not activate | Q86YC2 | PALB2 |
| Q8N163 | CCAR2 | does not inhibit | P51948 | BRCA1 |
| P38398 | BRCA1 | does not inhibit | P38398 | MNAT1 |
| P51587 | BRCA2 | does not activate | Q06609 | RAD51 |
| P38398 | BRCA1 | does not activate | Q13287 | NM1 |
| P67870 | CSNK2B | does not activate | P38398 | BRCA1 |
| Q9Y4A5 | TRRAP | does not activate | P38398 | BRCA1 |
| P38398 | BRCA1 | does not activate | P26038 | MSN |
| P38398 | BRCA1 | does not activate | Q99708 | RBBP8 |
| P38398 | BRCA1 | does not activate | Q09028 | RBBP4 |
| P38398 | BRCA1 | does not activate | Q9Y216 | NINL |
| P38398 | BRCA1 | does not activate | Q16576 | RBBP7 |
| Q15648 | MED1 | does not activate | P38398 | BRCA1 |
| P38398 | BRCA1 | does not activate | Q92769 | HDAC2 |
| P38398 | BRCA1 | does not inhibit | P31749 | AKT1 |
| P38398 | BRCA1 | does not activate with | 60934 | NBN |
| Q86YC2 | PALB2 | does not interact | P51587 | BRCA2 |
| P38398 | BRCA1 | does not activate | P84022 | SMAD3 |
| P38398 | BRCA1 | does not inhibit | Q8WXE1 | ATRIP |
| P38398 | BRCA1 | does not activate | Q14032 | BAAT |
| P38398 | BRCA1 | does not activate | Q6R327 | RICTOR |
| P38398 | BRCA1 | does not activate | P31749 | AKT1 |
| P23771 | GATA3 | does not activate | P38398 | BRCA1 |
| P00519 | ABL1 | does not activate | P38398 | BRCA1 |
| P38398 | BRCA1 | does not activate | P52701 | MSH6 |
| P38398 | BRCA1 | does not activate | P51587 | BRCA2 |
| P38398 | BRCA1 | does not activate | P49959 | MRE11A |
| P38398 | BRCA1 | does not activate | Q92878 | RAD50 |
| P38398 | BRCA1 | does not activate | P40692 | MLH1 |
| P38398 | BRCA1 | does not activate | P43246 | MSH2 |
| P38398 | BRCA1 | does not activate | P35241 | RDX |
| Q08211 | DHX9 | does not inhibit | P38398 | BRCA1 |
| P84022 | SMAD3 | does not inhibit | P38398 | BRCA1 |
| P38398 | BRCA1 | does not activate | P35222 | CTNNB1 |
| P38398 | BRCA1 | does not activate | Q9BX63 | BRIP1 |
| P38398 | BRCA1 | does not activate | Q13547 | HDAC1 |
| P38398 | BRCA1 | does not activate | P04637 | TP53 |
| Q86YC2 | PALB2 | does not activate | P51587 | BRCA2 |
| P09651 | HNRNPA1 | does not inhibit | P38398 | BRCA1 |
| P38398 | BRCA1 | does not activate | Q6UWZ7 | FAM175A |
| Q92830 | KAT2A | does not activate | P38398 | BRCA1 |
| P38398 | BRCA1 | does not activate | Q99759 | MAP3K3 |
| P38398 | BRCA1 | does not inhibit | Q86YC2 | PALB2 |
| Uniprot KB | Name | Displayed Name* |
| O14757 | Serine/threonine-protein kinase Chk1 | CHK1 |
| P03372 | Estrogen receptor | ESR1 |
| P06493 | Cyclin-dependent kinase 1 | CDK1 |
| P14635 | G2/mitotic-specific cyclin-B1 | CCNB1 |
| P14859 | POU domain, class 2, transcription factor 1 | PO2F1 |
| P16104 | Histone H2AX | H2AX |
| P30304 | M-phase inducer phosphatase 1 | MPIP1 |
| P30305 | M-phase inducer phosphatase 2 | MPIP2 |
| P30307 | M-phase inducer phosphatase 3 | MPIP3 |
| P31749 | RAC-alpha serine/threonine-protein kinase | AKT1 |
| P31947 | 14-3-3 protein sigma | 1433S |
| P38398 | Breast cancer type 1 susceptibility protein | BRCA1 |
| P40692 | DNA mismatch repair protein Mlh1 | MLH1 |
| P43246 | DNA mismatch repair protein Msh2 | MSH2 |
| P51587 | Breast cancer type 2 susceptibility protein | BRCA2 |
| P52701 | DNA mismatch repair protein Msh6 | MSH6 |
| Q09028 | Histone-binding protein RBBP4 | RBBP4 |
| Q13547 | Histone deacetylase 1 | HDAC1 |
| Q16576 | Histone-binding protein RBBP7 | RBBP7 |
| Q86YC2 | Partner and localizer of BRCA2 | PALB2 |
| Q92769 | Histone deacetylase 2 | HDAC2 |
| Q99708 | DNA endonuclease RBBP8 | COM1 |
| Q99728 | BRCA1-associated RING domain protein 1 | BARD1 |
| P04637 | Cellular tumor antigen p53 | P53 |
| Q03468 | DNA excision repair protein ERCC-6 | ERCC6 |
Supplemental data 3.Link Information: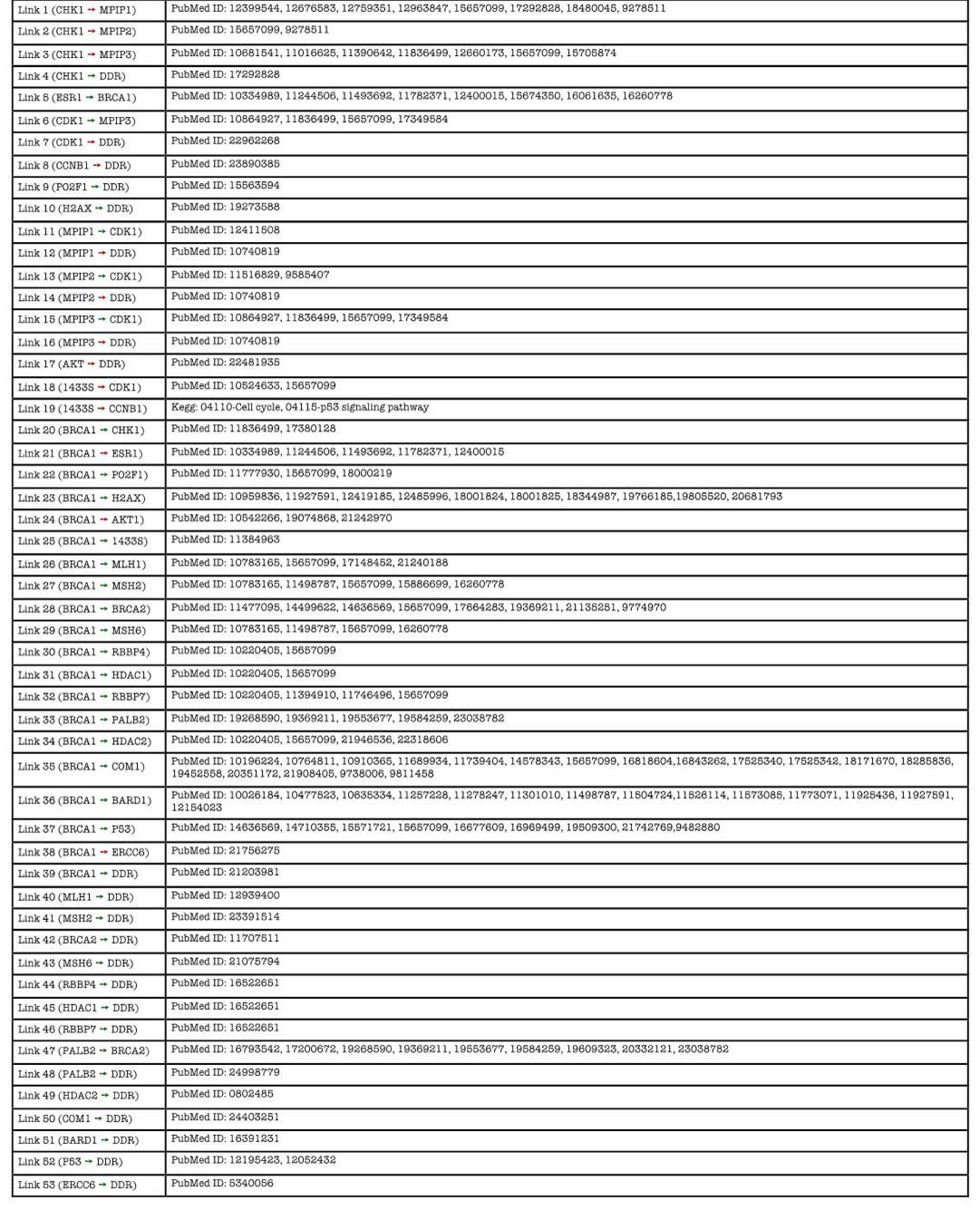
STIMULUS: Activation of BRCA1 A ➙B: Activation relationship
RESPONSE: DDR (DNA Damage Response)
A ➙B: Inhibition relationship
The model corresponding to the mutated variant has been defined with restrictions that eliminate functional associations that depend on the BRCT domain. Interestingly, the formation of the BRCA1 complexes A, B and C was impaired in the mutated variant model, since the interaction with Abraxas/FAM175A, Bach1/BRIP1 and CTIP proteins require the BRCT domain of BRCA1.
According to our results, DNA damage response is severely affected by the mutation (Figure 4), being the most relevant consequences.
Figure 4.Mechanistic comparison of the functional BRCA1 protein and the mutated BRCA1 protein. The description of the nodes and interactions is available as Supplementary data.
Mismatch repair (MMR): Since the mutated BRCA1 cannot activate MLH1, MSH2 or MSH6, which are key mediators of this repair mechanism, it is expected that MMR becomes severely impaired. As a result, mismatch mutations that are not repaired might persist and promote the apparition of tumors.
Homologous recombination (HR): BRCA2, COM1 and PALB3 are central mediators of the HR process, so their loss might result in the complete impairment of this pathway. Consequently, mutations that should be repaired by means of this process will not be fixed, potentially leading to cancer. HR participates in the repair of DNA double-stranded breaks (DSB) and interstrand crosslinks (CLS), as well as in providing support for DNA replication 26.
Nucleotide excision repair: Although the BRCA1-P53 interaction is lost, the inhibition of ERCC6, also known as Cockayne syndrome group (CSB) 27, appears to acquire a more prominent role. ERCC6 is known to promote the ubiquitination of P53 28, which is considered by some authors as a “nucleotide excision repair master-switch”. Hence, by inhibiting ERCC6, the BRCA1 variant may stabilize the activity of P53 induced by alternative routes. ERCC6 is crucial for transcription-coupled DNA repair (TCR), which removes the cytotoxic transcription-blocking DNA lesions 29, by recruiting the components of the NER machinery such as RNAPIIo or XAB2 30.
Non-homologous end joining (NHEJ): With BRCA1 unable to activate MSH6, a certain impact on this process is expected. However, H2AX is still active, as well as other proteins that are important for this mechanism. Hence, little real change in this mechanism is expected.
Cell cycle arrest: Despite the disruption of the interactions with key proteins of the cell cycle, namely AKT, P53, HDAC 1/2 and RBBP 4/7, the Pro1821Ala variant is still capable of binding and affecting other mediators of cell cycle arrest. Thus, albeit this process will be much more fragile and sensitive to future mutations, it is expected that BRCA1 retains a certain ability to stop the cell cycle.
The different biological processes and protein interaction altered by the studied mutation are summarized in Table 1.
Table 1. Summary of the differences in DNA damage repair related processes between wild-type and mutated BRCA1.| Process | Affected | Not affected | New |
| MMR | MLH1, MSHS, MSH6 | None | - |
| HR | COMI, BRCA2, PALB2 | BARDI, H2AX | - |
| NER | P53 | None | ERRC6 |
| NHEJ | MSH6 | H2AX | - |
| Cell cycle | P53, AKT1, HDAC1, HDAC2, RBBP4, RBBP7 | COMI | MPIP1, MPIP2, MPIP3, 1433S, CDK1, CCNB1 |
Overall, the comparison between the MoAs of the full-length BRCA1 and its mutated variant shows that the latter undergoes a drastic reduction in its ability to modulate a number of proteins and pathways which are critical for DNA repair and cell cycle control.
Discussion
In order to investigate the hypothetically pathogenic role of the BRCA1 c.5434C->G (p.Pro1812Ala) mutation, a study was carried out using computational methods. A structural model was created and it was assessed from a functional and systems biology point of view.
The c.5434C->G (p.Pro1812Ala) mutation causes an important splicing defect in BRCA1, resulting in the omission of exon 23, which in turn leads to the appearance of a premature stop codon (p.Gly1803GlnfsX11) 10. However, and according to our results, it does not lead to secondary or tertiary structural changes. The structural configuration of the wild-type protein is preserved, which coincides with the findings of Drikos et al.: the c.5434C->G (p.Pro1812Ala) mutation preserves the ability to interact through the BRCT domain with proteins such as BRIP1 and RBBP8. But binding affinity and stability were substantially modified, which could alter the conformational equilibrium and therefore affect the protein's function in vivo 11.
As a result of a dysfunctional BRCT domain, the mutated protein cannot carry out its role in DNA damage response and cell cycle regulation.
BRCA1 activity that depends on proteins that interact with other domains, such as interaction of the RING domain with the BARD1 protein, is not expected to be affected by this mutation 31.
In the c.5434C->G (p.Pro1812Ala) variant, p53 is not activated by the altered BRCT domain. The initiation of the apoptotic response may therefore be compromised 24. Although this process will be much more fragile and sensitive to future mutations, it is expected that BRCA1 will retain a certain ability to stop the cell cycle. The activation of p53 and other key mediators of the cell cycle and apoptosis does not depend on BRCA1 alone.
The results of structural and mechanical analyses mean that the c.5434C->G (p.Pro1812Ala) variant can be reclassified from a variant of unknown significance to a pathogenic variant. The following evidence supports this statement:
Segregation: the c.5434C->G (p.Pro1812Ala) variant was suspected of being harmful in at least one family 32.
Population Frequency: The c.5434C->G (p.Pro1812Ala) mutation has been identified in patients with breast cancer or ovarian cancer, in both Spanish and non-Ashkenazi Jewish women 32.
Evolutionary Preservation: At position 1812 of the BRCA1 protein the amino acid proline is highly conserved between species, suggesting that the normal function of the protein plays an essential role. The c.5434C->G (p.Pro1812Ala) variant causes a change in the splicing pattern of BRCA1 10. Transcriptions expressed from the mutant c.5434C->G (p.Pro1812Ala) exhibit an omission of exon 23 in 75% of cases.
Functional assay: Our study did not indicate any significant change in structure. However, Drikos et al. observed a lower thermal stability that could influence conformational equilibrium. Assays of c.5434C->G (p.Pro1812Ala) mutation activity showed slightly reduced gene activity 11. An in vivo reporter gene assay showed that in c.5434C->G (p.Pro1812Ala) mutation activity clearly affects function 32. This assay assessed the BRCT domain's ability to induce transcriptional activation 10.
BRCA1 mutations with a similar splicing effect, including the neighbouring variants c.4987-3C>G 33 and c.5075-1G>C 34, were classified as pathogenic. Another study that examined a missense variant of unknown significance in the C-terminus region, close to variant c.5434C->G (p.Pro1812Ala), revealed significant alterations in BRCT structure and function 35.
Systems biology tools were used to evaluate the impact of the BRCA1 c.5434C->G (p.Pro1812Ala) mutation on DNA damage response from a mechanistic perspective. The results of this analysis support the findings of structural and functional methods explaining how the c.5434C->G (p.Pro1812Ala) mutation leads to impaired DNA repair and cell cycle control. This conclusion can be extrapolated to all mutations that cause loss of BRCT domain activity.
As regards DNA repair pathways, Mismatch Repair (MMR) and homologous recombination can be strongly altered in cells that harbour the c.5434C->G (p.Pro1812Ala) variant, whereas the effect on non-homologous end joining (NHEJ) and nucleotide excision repair (NER) seem to be less significant. Each of these mechanisms plays an independent and irreplaceable role in DNA repair. Failure of any one of them makes it impossible to repair a certain type of damage, which will eventually lead to an accumulation of mutations.
Together with structural and functional analysis, mechanistic insight provides a sound basis for defining the c.5434C->G (p.Pro1812Ala) mutation as a pathogenic variant. The BRCA1 c.5434C->G (Pro1812Ala) mutation causes deletion of the second BRCT domain, which plays a key role in critical processes, such as DNA repair and cell cycle control. It is therefore to be expected that this variant will not be able to exert its tumour suppressor action.
In conclusion, the c.5434C->G (p.Pro1812Ala) mutation creates an environment with increased risk of binding and tolerance to damage, which promotes the accumulation of mutations. Despite some compensation mechanisms, DNA damage whose repair is not induced, cell cycle arrest or stimulation of the apoptotic machinery may go unnoticed. This phenomenon can lead to mutations in other tumour suppressor genes or oncogenes, which causes cancer to develop. This hypothesis is consistent with Knudson's two-hit hypothesis 36 and with increased cancer susceptibility associated with BRCA1 37.
Acknowledgements
“The research leading to these results has received funding from the European Union's Seventh Framework Programme for research, technological development and demonstration under grant agreement No. 306240.”
References
- 1.Milne R L, Osorio A, TRY Cajal, Vega A, Llort G et al.(2008May1) The average cumulative risks of breast and ovarian cancer for carriers of mutations in BRCA1 and BRCA2 attending genetic counseling units in Spain. Clin Cancer Res [Internet].[cited2015Feb10];.Availablefrom:http://www.ncbi.nlm.nih.gov/pubmed/18451254. 14(9), 2861-9.
- 2.Lorick K L, Jensen J P, Fang S, Ong A M, Hatakeyama S et al.. (Sep28,1999). RING fingers mediate ubiquitin-conjugating enzyme (E2)-dependent ubiquitination.Proc Natl Acad Sci U S A[Internet].[cited2015Jan17].Availablefrom:http://www.pubmedcentral.nih.gov/articlerender.fcgi?ar-tid=18039&tool=pmcentrez&rendertype=abstract 96(20), 11364-9.
- 3.Wang B, Matsuoka S, Ballif B A, Zhang D, Smogorzewska A et al.(2007May25) Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science [Internet].[cited2016Nov20]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/17525340. 316(5828), 1194-8.
- 4.Kim H, Huang J, Chen J.(Aug2007) CCDC98 is a BRCA1-BRCT domain-binding protein involved in the DNA damage response. Nat Struct Mol Biol [Internet]. [cited 2015Mar6]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/17643122. 14(8), 710-5.
- 5.Yu X, CCS Chini, He M, Mer G, Chen J. (2003) The BRCT domain is a phospho-protein binding domain. Science [Internet]. [cited2015Mar6]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/14576433 302(5645), 639-42.
- 6.Yu X, Chen J.(Nov2004) DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domains. Mol Cell Biol [Internet].[cited2015Mar6];Availablefrom:http://www.pubmedcentral.nih.gov/articlerender.fcgi?a-tid=522253&tool=pmcentrez&rendertype=abstract. 24(21), 9478-86.
- 7.Wang B.(2012Feb27) BRCA1 tumor suppressor network: focusing on its tail. , Cell Biosci[Internet].[cited2017Jan11];Availablefrom:http://cellandbioscience.biomedcentral.com/articles/10.1186/2045-3701-2-6 2(1), 6.
- 8.MSY Huen, SMH Sy, Chen J.BRCA1 and its toolbox for the maintenance of genome integrity. Nat Rev Mol Cell Biol [Internet]. [cited2015Jan14]; Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?atid=3899800&tool=pmcentrez&rendertype=abstract. 11(2), 138-48.
- 9.Joosse S A.(May2012) BRCA1 and BRCA2: a common pathway of genome protection but different breast cancer subtypes. Nat Rev Cancer [Internet].[cited2015Mar6] author reply372.Availablefrom:http://www.ncbi.nlm.nih.gov/pubmed/22525577. 12(5), 372.
- 10.Gaildrat P, Krieger S, Théry J-C, Killian A, Rousselin A et al.(Jun2010) The BRCA1 c.5434C->G (p.Pro1812Ala) variant induces a deleterious exon 23 skipping by affecting exonic splicing regulatory elements. J Med Genet [Internet]. [cited2015Feb8]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/20522429. 47(6), 398-403.
- 11.Drikos I, Nounesis G.Vorgias CE. (2009Nov1) Characterization of cancer-linked BRCA1-BRCT missense variants and their interaction with phosphoprotein targets. Proteins[Internet].[cited2015Mar6]Availablefrom:http://www.ncbi.nlm.nih.gov/pubmed/19452558 77(2), 464-76.
- 12.Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S et al.CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. , J Comput Chem[Internet][cited2017Jan11]:NA-NA.Availablefrom:http://www.ncbi.nlm.nih.gov/pubmed/19575467 31(4).
- 13.Herrando-Grabulosa M, Mulet R, Pujol A, Mas J M, Navarro X et al.. (2016Jan25) Novel Neuroprotective Multicomponent Therapy for Amyotrophic Lateral Sclerosis Designed by Networked Systems. Duce JA, editor. PLoS One[Internet].[cited2017Jan11];Availablefrom:http://dx.plos.org/10.1371/journal.pone.0147626 11(1), 0147626.
- 14.Gilson M K, Liu T, Baitaluk M, Nicola G, Hwang L et al.(Jan4,2016)BindingDB in2015:A public database for medicinal chemistry, computational chemistry and systems pharmacology.Nucleic Acids Res[Internet].[cited2017Jan11]; Availablefrom:https://academic.oup.com/nar/article-lookup/doi/10.1093/nar/gkv1072. 44-1.
- 15.Chatr-Aryamontri A, Breitkreutz B-J, Oughtred R, Boucher L, Heinicke S et al.(2015Jan28) The BioGRID interaction database: 2015 update. Nucleic Acids Res [Internet].[cited2017Jan11].43(Databaseissue):D470-8.Available from: https://academic.oup.com/nar/article-lookup/doi/10.1093/nar/gku1204.
- 16.Kerrien S, Aranda B, Breuza L, Bridge A, Broackes-Carter F et al.(2012Jan1) The IntAct molecular interaction database in 2012. Nucleic Acids Res[Internet].[cited2017Jan11].40(Databaseissue):D841-6.Availablefrom:http://nar.oxfordjournals.org/lookup/doi/10.1093/nar/gkr1088.
- 17.Fabregat A, Sidiropoulos K, Garapati P, Gillespie M, Hausmann K et al.(2016Jan4) The Reactome pathway Knowledgebase. Nucleic Acids Res[Internet].[cited2017Jan11];44(D1):D481-7.Availablefrom: https://academic.oup.com/nar/article-lookup/doi/10.1093/nar/gkv1351.
- 18.Milacic M, Haw R, Rothfels K, Wu G, Croft D et al.(2012Nov8) Annotating cancer variants and anti-cancer therapeutics in reactome. , Cancers(Basel)[Internet].[cited2017Jan11].Availablefrom:http://www.mdpi.com/2072-6694/4/4/1180/ 4(4), 1180-211.
- 19.Edgar R, Domrachev M, Lash A E.(2002Jan1) Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. , Nucleic Acids Res[Internet][cited2017Jan11].Availablefrom:http://www.ncbi.nlm.nih.gov/pubmed/11752295 30(1), 207-10.
- 20.Barrett T, Wilhite S E, Ledoux P, Evangelista C, Kim I F et al. (2013) NCBI GEO:archive for functional genomics data sets--update.Nucleic Acids Res[Internet].[cited2017Jan11];41(Databaseissue):D991-5.Availablefrom:https://academic.oup.com/nar/article-lookup/doi/10.1093/nar/gks1193.
- 21.Wishart D S, Knox C, Guo A C, Shrivastava S, Hassanali M et al.(2006Jan1).DrugBank: a comprehensive resource for in silico drug discovery and exploration.Nucleic Acids Res[Internet].[cited2017Jan11];34(Databaseissue):D668-72.Availablefrom:https://academic.oup.com/nar/article-lookup/doi/10.1093/nar/gkj067.
- 22.Bishop C M. (2004) . Pattern Recognition and Machine Learning.Jordan,M,Kleinberg,J,Schölkopf B,editors.NY,USA:Springer .
- 23.Perera S, Artigas L, Mulet R, Mas J M, Sardón T. (2014) Systems biology applied to non-alcoholic fatty liver disease (NAFLD): treatment selection based on the mechanism of action of nutraceuticals. , Nutrafoods[Internet].Availablefrom:http://dx.doi.org/10.1007/s13749-014-0022-5 13(2), 61-8.
- 24.. BRCA1_HUMAN. UniProtKb: P38398.2 [Internet]. 2015 [cited 2015 Feb 10]. Availablefrom:http://www.ncbi.nlm.nih.gov/protein/P38398 .
- 25.TMA Abdel-Fatah, Perry C, Arora A, Thompson N, Doherty R et al.(2014Dec1) Is there a role for base excision repair in estrogen/estrogen receptor-driven breast cancers? Antioxid Redox Signal[Internet].[cited2015Feb10];Availablefrom:http://www.ncbi.nlm.nih.gov/pubmed/25111287. 21(16), 2262-8.
- 26.Li X, Heyer W-D.. (Jan2008) Homologous recombination in DNA repair and DNA damage tolerance.Cell Res[Internet].[cited2017Jan15]Availablefrom:http://www.nature.com/doifinder/10.1038/cr.2008.1 18(1), 99-113.
- 27.Vélez-Cruz R, Egly J-M.(May2013) Cockayne syndrome group B (CSB) protein: at the crossroads of transcriptional networks. Mech Ageing Dev [Internet].[cited2017Jan15];134(5–6): 234–42.Availablefrom:http://linkinghub.elsevier.com/retrieve/pii/S0047637413000419.
- 28.Latini P, Frontini M, Caputo M, Gregan J, Cipak L et al.. (Nov1,2011) CSA and CSB proteins interact with p53 and regulate its Mdm2-dependent ubiquitination. Cell Cycle[Internet].[cited2017Jan15];Availablefrom:http://www.tandfonline.com/doi/abs/10.4161/cc.10.21.17905 10(21), 3719-30.
- 29.Boom V van den, Citterio E, Hoogstraten D, Zotter A, Egly J-M et al.. (Jul5,2004) DNA damage stabilizes interaction of CSB with the transcription elongation machinery.J Cell Biol[Internet].[cited2017Jan15];Availablefrom:http://www.jcb.org/lookup/doi/10.1083/jcb.200401056 166(1), 27-36.
- 30.Fousteri M, Vermeulen W, van Zeeland AA, LHF Mullenders.(Aug2006) Cockayne syndrome A and B proteins differentially regulate recruitment of chromatin remodeling and repair factors to stalled RNA polymerase II in vivo. Mol Cell [Internet].[cited2017Jan15];Availablefrom:http://linkinghub.elsevier.com/retrieve/pii/S1097276506004655. 23(4), 471-82.
- 31.Wu L C, Wang Z W, Tsan J T, Spillman M A, Phung A et al.(Dec1996) Identification of a RING protein that can interact in vivo with the BRCA1 gene product.Nat Genet[Internet].[cited2015Mar6];Availablefrom:http://www.ncbi.nlm.nih.gov/pubmed/8944023. 14(4), 430-40.
- 32.Kaufman B, Laitman Y, Carvalho M A, Edelman L, Menachem T D et al.. (Jan2006) The P1812A and P25T BRCA1 and the 5164del4 BRCA2 mutations: occurrence in high-risk non-Ashkenazi Jews.Genet Test[Internet].[cited2015Mar6];Availablefrom:http://www.ncbi.nlm.nih.gov/pubmed/17020472. 10(3): 200 – 7 .
- 33.Brandão R D, van Roozendaal KEP, Tserpelis D, Caanen B, Gómez García E et al. (2012) c.4987-3C>G is a pathogenic mutation. Breast Cancer Res Treat[Internet].[cited2015Feb8];Availablefrom:http://www.pubmedcentral.nih.gov/articlerender.fcgi?atid=3249558&tool=pmcentrez&rendertype=abstract. 131(2), 723-5.
- 34.Steffensen A Y, Dandanell M, Jønson L, Ejlertsen B, Gerdes A-M et al.(Dec2014) Functional characterization of BRCA1 gene variants by mini-gene splicing assay. , Eur J Hum Genet[Internet].[cited2015Jan30]; Availablefrom:http://www.pubmedcentral.nih.gov/articlerender.fcgi?a-tid=4231409&tool=pmcentrez&rendertype=abstract 22(12), 1362-8.
- 35.Quiles F, Fernández-Rodríguez J, Mosca R, Feliubadaló L, Tornero E et al.. (Jan2013) Functional and structural analysis of C-terminal BRCA1 missense variants. PLoS One[Internet].[cited2015Mar6];Availablefrom:http://www.pubmedcentral.nih.gov/articlerender.fcgi?a-tid=3629201&tool=pmcentrez&rendertype=abstract 8(4), 61302.
Cited by (3)
This article has been cited by 3 scholarly works according to:
Citing Articles:
CPT Pharmacometrics & Systems Pharmacology (2023) OpenAlex
CPT: Pharmacometrics & Systems Pharmacology (2023) Crossref
Valentin Junet, Pedro Matos-Filipe, J. M. García-Illarramendi, Esther Ramírez, B. Oliva et al. - CPT: Pharmacometrics & Systems Pharmacology (2023) Semantic Scholar
Journal of Cancer Genetics and Biomarkers (2017) OpenAlex