
Molecular Analysis of 6-pyruvoyltetrahydropterin Synthase Gene in Atypical Phenylketonuric Egyptian Patients
Abstract
Background
Hyperphenylalaninemia (HPA) combined with neurological signs due to impaired catecholamine, dopamine and serotonin synthesis. Symptoms may appears in first week of life but most seen in age of 4 months. Atypical PKU disease caused mainly by deficiency in 6-pyruvoyltetrahydropterin synthase (PTPS) involved in synthesis of BH4. Clinical symptoms may include poor sucking, impaired tone, ataxia, and seizures. The purpose of this study was to analyze the genotype-phenotype relation among BH4 deficient patients because of PTPS mutations in different state of Egypt.
Methods
Suspected PKU patients loaded with phenylalanine/Kuvan, and the level of phe and phe/tyrosine ratio determined using tandem mass spectrometry by dried blood spots. Blood samples of 13 unrelated Egyptian patients were collected for total RNA extraction, amplification of PTPS gene by PCR followed with sequencing by Sanger method and finally mutations were recorded for genetic analysis.
Results
The mean value of phe in 13 patients decreased after loaded of phenylalanine from 482.5μmol/L to 270.63 μmol/L as well as phe/tyrosine ratio was decreased from 13.4 to 6.36 after 24hour of treatment with Kuvan. Sanger sequencing of PTPS gene of those patient showed 21 SNPs and Indels mutations. The most repeated mutation is a novel 23 base pair homozygous deletion in 12/13; c.200C>T in four patients, a novel c.86A>T in two patients and three different mutations located once in three different patients (novel c.22C>T; novel c.273G>A and 405T>C) among patients. On amino acid predicted sequences 4 different types of mutations on protein level were presented, 1 deletion mutation in seven amino acid and 3 different missense mutations in addition to 2 silent mutations among 13 patients.
Conclusion
Patients were the first case of clinical diagnosis as hyperphenylalaninemia (HPA) undergoing genetic diagnosis for PTPS deficiency in Egypt. The sever HPA patients with severe nervous system damage mainly accompanied with deletion mutations and should pay more attention to the BH4 deficiency. While mild HPA is associated with base substitution mutations with mainly transition mutations (7/9; 78%). Next-generation sequencing technique can increase the mutation detection rate when the hereditary diseases are highly suspected in clinic.
Author Contributions
Academic Editor: Pengyang Li, B.S. in Materials Science & Engineering, University of Illinois at Urbana-Champaign, M.S. in Materials Science & Engineering, Stanford, United States.
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2020 Ahmed F. Mohamed, et al.
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Introduction
Phenylketonuria (PKU; OMIM#261600) is autosomal recessive metabolic disorder disease, which is characterized by a disruption in the ability to metabolize amino acid phenylalanine (phe) into tyrosine (tyr) that is a precursor form of dopamine and other catecholamines. Individuals with PKU have deficiency or reduced activity of the hepatic phenylalanine 4- hydroxylase (PAH; EC 1.14.16.1) enzyme, which is necessary for this metabolic process to occur and consequently accumulate serum levels of phe and decrease neurotransmitter production specially tyrosine level 1. The excess phe further competes with the available tyrosine and other large neutral amino acids (LNAAs) to cross the blood–brain barrier.
Abnormal signs and symptoms of PKU and BH4-deficiency patients include poor sucking, impaired tone, progressive intellectual disability, mousy odor, microcephaly, mental and growth retardation, hypotonia, and epilepsy 2. Frequent symptoms of PTPS deficiency, the most common form of BH4 deficiency resemble Parkinson disease characterized by a lake of dopamin in the basal ganglia 3. Other signs include characteristic truncal hypotonia, increased limb tone, postural instability, hypokinesia, dystonic limb movements, gait difficulties, hyper-salivation due to swallowing difficulties, drowsiness and irritability 4.
PKU is the most studied disease among inborn disorders of amino acid metabolism. The average incidence of PKU disease is about one per 10,000 newborns worldwide and varies widely around the world. PKU prevalence is as high as 1 in 2600 births in Turkey 5 to fewer than 1 in 100,000 births in Japan 6. Other reported incidences are 1 in 4500 births in Ireland 7, 1 in 10,000 births in the United Kingdom 8, 1 in 11,000 births in China 9. In addition, incidence in Thailand is 1 in 200,000 in newborns 10, incidence in Caucasians is approximately 1 in 10,000 births 11 and 1 in 15,000 births in the United States 12.
Infants with high level of phe may be due to PAH defect or BH4 deficiency usually appear normal at birth, and untreated infancy showed a continuous increasing in blood Phe concentration (hyperphenylalaninemia; HPA) and symptoms of PKU appeared, which categorized into a classical class and atypical class. The majority of worldwide PKU patients (98%) were classified as classic PKU, which is caused by mutations in PAH gene leading to absent or deficiency in PAH enzyme activity. About 2% of PKU cases 13 are BH4-deficient, caused by mutations in one or more of the five genes involved in BH4 biosynthesis; of 6-pyruvoyl-tetrahydropterin synthase (PTPS), GTP cyclohydrolase I (GCH1), quinoid dihydropteridine reductase (QDPR), pterin-4-alpha-carbinolamine dehydratase 1 (PCBD1), and sepiapterin reductase (SPR) 14 that caused a defect in the synthesis or regeneration of PAH cofactor (BH4). The latter requires supplementation of BH4 and various BH4-dependently synthesized neurotransmitters 15.
Atypical phenylketonuric patients distinguished from classical Phenylketonuric patients with specific diagnostic criteria (i) blood Phe concentration was slightly elevated; 4-20 mg/100ml; (ii) greater tolerance to dietary phenylalanine on loading BH4 test; (iii) urinary pterin analysis showed no significant abnormalities 16. A BH4 loading test should be performed to determine whether patients are BH4-responsive or unresponsive. Patients showed that BH4 significantly lowered serum phe concentrations in the majority of atypical PKU patients with mild hyperphenylalaninemia (MHPA), while no patients with classic phenylketonuria (PKU) had a response 17.
It should be noted that it is not easy to perform a BH4 loading test in patients > 2years who are well managed by diet and thus have low blood phe concentration and prefer to perform the test during the neonatal period when phe level is naturally elevated. Our team group recommended to use a BH4 loading test and treatment trial with sapropterin dihydrochloride (Kuvan®, BioMarin, CA, USA and Merck, Geneva, Switzerland) instead. It is a synthetic formulation of the 6R-isomer of BH4 that reduced blood phenylalanine (Phe) levels in patients with HPA due to tetrahydrobiopterin- (BH4) responsive PKU by activating residual PAH enzyme activity in addition to improve the normal oxidative metabolism of phe of all ages and has been commercially available for several years. This agent was authorized and approved for the treatment of HPA due to BH4 responsiveness PKU in pediatric and adult patients without age restriction in 2007 in the USA and older than 4 years in 2008 in Europe 18. Kuvan works in the same way as BH4 metabolized and was used in conjunction with or without a Phe- restricted diet. Treatment with Kuvan can activate residual PAH and decrease phe levels in BH4 responsiveness PKU patients 19. The effectiveness of Kuvan treatment has been demonstrated in short-term and long-term studies 20 included patients aged 6 months–5 years and shown the best effectivness of treatment in this age range 21.
The PTPS gene encodes 6-pyruvoyl-tetrahydropterin synthase (EC 4.6.1.10), an enzyme involved in the second step of de novo biosynthesis of tetrahydrobiopterin (BH4) which is composed of a pair of trimers arranged in a head-to-head fashion to form the functional hexamer. The homohexamer contains six active sites that are located on the interface of three monomers, two subunits from one trimer and one subunit from the other trimer 22, 23. The PTPS gene is located from base 112,226,364 to base 112,233,972 on chromosome 11 spans about 8 kb and consists of 6 exons 24 coding a polypeptide of 145 amino acids.
The respond to the dietary treatment of classical PKU and BH4- deficiency patients is different. A life-long Phe restricted diet with or without BH4 has been the only possible treatment for classical PKU patients, this diet therapy is highly recommended to continue throughout their life to prevent behavioral disorders, cognitive, and emotional dysfunction 25. On the other hand, PKU patients with BH4 deficiency require BH4 supplement, generally 2–20 mg/kg/day 26, 27. Treatment has proved to be very effective in preventing the devastating consequences of PAH deficiency when started early in life (neonatal stage). The aim of this work is to find the phenotype/genotype correlation in atypical PKU between the type of mutations in PTPS gene and clinical diagnosis.
Materials and Methods
Patients
This study carried out on selected 13 Egyptian patients of unrelated families with mild hyperphenylketonuria (MHPA; >200<1200 μmol/L), they were positive for performing the BH4 loading test to be atypical PKU patients to determine the mutation/s in PTPS gene. Patients diagnosed and followed up at Genetics Unit, Pediatric hospital, Ain Shams University from January 2017 to October 2018. They were 7 males and 6 females aged from 2 month to 6 years suffering from poor feeding, delayed motor, mental development and coma. Most of these patients were identified during neonatal screening program.
Selected patients diagnosed and confirmed as MHPA or atypical PKU by measuring the level of phe and phe/tyrosine ratio before and after BH4 analogous loading test by tandem mass. In which, dried blood level of phe concentration at a diagnosis time of selected PKU patients is ≥200 μmol/L and 1200 μmol/L and phe/tyrosine ratio > 3 obtained on two separate samples. They also were positive response to BH4 analogous loading test (the initial dried blood phe level was decreased by at least by 40% after 12 h following one dose of 20 mg/kg Kuvan challenge).
Loading Kuvan test carried out according to the standard protocol of Ain Shams Hospital. In a test dried blood samples were collect on fasting for at least 8 hours, then patients orally received phenylalanine dissolved in breast milk or water at dose of 100 mg/kg body weight. Baseline dried blood Phe level was measured after 3 hr of receiving phe suspension and at that time, the patient treated with 20 mg/kg body weight of Kuvan dissolved also in breast milk. Following Kuvan administration and throughout the entire test, a normal breast milk or regular infant formula-feeding regimen must be provided on demand and Phe-free medical formula must not be given. Dried blood samples were taken at time 4, 8, 12 and 24 hours after Kuvan administration. The test its results must be available over a period of 24 h to avoid delaying treatment of neonates with classical PKU for longer than 24 h. All blood samples used to measure the concentration of phenylalanine and phe/tyrosine ratio as well by tandem mass from either venous blood or dry blood spot.
Patients’ parents/guardians or one of his relatives gave written signed informed consent for participation in the study before any trial related procedures were performed and had to be willing to comply with maintain strict adherence to the diet. This study approved by the Research Ethics Committee (REC) of Medical Hospital Staff of Faculty of Medicine, Ain Shams University (FWA000017585), for experiments involving humans.
Dry Blood Spots (DBSs) Test by LC/MS-MS
Dry blood samples were taken from patients by heel stick, spotted on Whatman filter paper cards (Schleicher and Schuell 903;Dassel, Germany). They left to dry before screening by tandem mass spectrometry (LC/MS-MS; ACQUITY UPLC ®) to determine the level of phe and phe/tyrosine ratio in their blood on normal diet and throw loading test
Blood Samples
Blood samples were collected from patients in potassium EDTA vacutainer test tubes. RNA extraction and purification was carried out in the same day of sample collection by using GeneJETᵀᴹ whole blood RNA purification Mini Kit, #K0761 (Thermoscientific) according to the manufacturer’s protocol. CDNA was synthesized by QuantiTect Reverse transcription Kit (QIAGEN; cat. no. 205314). PCR was carried out by Go Taq® green master mix, 2X, (Promega), cat. no. #M7112. One set of primers (forward and reverse) were designed for amplification of specific sits covering entire CDs length of mRNA of PTPS gene, they were designed by using web based primer-blast tool, NCBI (National center for biotechnology information). The forward prime sequence is 20 bases starting at position 76 with sequence 5`- GAAGATGAGCACGGAAGGTG-3` and reverse primer is 20 bases at position 606 with sequence 5`- ACGTGTTGACCTCTTAATAT-3` spanning amplicon of 512 base pair. PCR was carried out on the Gene Amp PCR system 9700 (Applied Biosystems, CA). The PCR cycles were carried out as follows: 5 minutes initial denaturation at 94°C followed by 30 cycles of amplification, each including 15 seconds denaturation at 94°C, 30 seconds annealing at 58°C, and 45 seconds extension at 72°C. Amplification cycles followed by 5 minutes final extension at 72°C and then cooled to 4°C. The amplification product was separated by agarose gel electrophoresis to confirm the size of amplicons.
Sequencing of PCR Amplicon
Bidirectional sequencing of the purified PCR product in both directions (GATC Company, Germany) using ABI 3730XL DNA sequencer (Applied Biosystem, USA) by using forward and reverse primers. Sequencing results are used for nucleotide blast on http://blast.ncbi.nlm.nih.gov/Blast.cgi (National center for Biotechnology) to make alignment with the PTPS gene normal strand by using its accession number NM_000317.3 to detect new mutations. All homozygous and compound heterozygous variants were included.
All detected mutations submitted on https://mutalyzer.nl/name- checker and polyphen2 software to detect the effect of the mutation on the amino acids sequence of the PTPS peptide and detect the type and degree of pathogenicity of mutations. The mutalyzer gives also the expected protein sequence of the missense, nonsense and frame shift mutations. The amino acid sequences submitted to http://raptorx.uchicago.edu/ to detect the expected 3D protein structure of the mutant enzyme and compare it with the normal PTPS. The novel mutations detected from this study were submitted to GenBank database.
Analysis of Data
Sequence results used for nucleotide blast by the online program http://blast.ncbi.nlm.nih.gov/Blast for alignment with normal PTPS gene to detect a mutations. The three major databases for known pathogenic and likely pathogenic mutations used including ClinVar, OMIM, and HGMD program.
In total, variants of patients were generated and processed to select the candidate pathogenic mutations in coding regions (missense, nonsense, coding indels and frameshift) which were predicted to be neutral, deleterious or damaging by many software programs. Two software to determine the 3D structure and active sites; RaptorX (web server predicting protein structure property solely based on protein sequence or sequence profile) and I- TASSER (Iterative Threading ASSEmbly Refinement). Three software predicting pathogenicity; Sorting intolerant from tolerant (SIFT) (J. Craig Venter Institute, San Diego, CA, USA), protein variation effect analyzer (PROVEAN), and polymorphism phenotyping v2 (PolyPhen-2) (Division of Genetics, Brigham & Women’s Hospital, Harvard Medical School, Boston, MA, USA), were applied to predict the effects of mutations on the protein structure and function. In addition, SPSS version 22 software used for statistical analysis of means ±SD
Results
This study was carried out on 13 patients (7 males and 6 females) from unrelated families (Table 1) classified as atypical PKU in the Genetics Research Unit, Pediatrics Hospital, Ain Shams University. Most of these patients identified during neonatal screening program. They distributed through different ten governmental areas in Egypt and suffering from PKU symptoms and milestones. Patients were stratified according to age, where 4 patients were <12 months, 5 patients were 12 to <36 months, and 4 patients were 36 to <72 months. The mean ±SD of age at diagnosis was 26.7±11.3 month and age at onset of the disease is 3±0.05 month. One female patient died during the treatment with Kuvan. Severity of PKU-BH4 responsiveness patients were classified as mild HPA-PKU in which the concentration of phe at baseline (at 3 h after administration of 100 mg/kg body weight phe) < 1,200 μmol/L (12 patients; 92%) and severe PKU where the phe concentration at baseline >1,200 μmol/L (one patient; 8%).
Table 1. Demographic characteristics of MHPA patients| characteristics | |
| Gender n (%) | |
| Males | 7 (54) |
| females | 6 (46) |
| Age, months | |
| mean± SD | 26.7±11.3 |
| min; max | 2-72 |
| Age group, n (%) | |
| <12 months | 4 (30.7) |
| 12<36 | 5 (38.6) |
| 36-72 | 4 (30.7) |
| Onset of the disease, months | |
| mean± SD | 3±0.05 |
| min; max | 3-4 |
| PKU severity n (%) | |
| Mild | 12 (92) |
| Severe | 1 (8) |
| Mortality state n (%) | |
| Died | 1 (7.7) |
| Lived | 12 (92.3) |
Symptoms and Milestones Before Kuvan Treatment
Almost all patients were suffering from global developmental delay noticed at age of 3 month as most of them could not support neck or recognize parents and other 3 cases (p 7, p 8, p 11) were suffering from convulsions at age of 6 month. They suffering from hypotonia, briskly reflexes or elicited reflexes, except (P11) suffering from mild hypotonia in lower limbs, elicited reflexes and (P1) was with normal tone and normal reflexes. Only one patient (P 12) site lone and recognize families, all other patients were suffering from uncontrolled support neck or recognize family, (P2) was suffering from global developmental delay noticed at age of 3 month.
Biochemical Analysis
First blood samples were collected from patient fasting for at least 8 hours, just after collecting fasting sample patients take phenylalanine powder dissolved in water or breast milk (by dose of 100 mg/Kg body weight). Three hour later of phenylalanine administration the second sample was taken, immediately patients administrated once with Kuvan (powder or tablets) at dose of 20mg/kg body weight dissolved in water or apple juice for infant or soluble in breast milk for neonates. After loaded of Kuvan 4 samples were collected at 4, 8, 12 and 24 hours. All blood samples used for measuring phe concentration in umol/l and phe/tyrosine ratio by tandem mass with taking care of clinical behavior during this period.
Milestones After Treatment with Kuvan
Patients (P4, 5, 11, 10, 13) could support neck and recognize families, (P2) recognize mother, follow objects; (P3) can sit alone, crawl, recognize parents and say 2 words. Other patient (P11) can stand supported, recognize family, say one word, (P8) mild neck support, can follow objects, (P7) can walk upstairs alone, can say sentence of 3 words, can say 150 words, (P12) can walk supported, (P9) can sit alone and recognize mother, (P1, 6) can walk alone, say one word, recognize family.
The tandem mass spectrometry (LC/MS/MS) result of PKU-BH4 responsiveness patients showed that phe concentration and phe/tyrosine ratio on fasting were highly elevated reaching to 227.8±51.1μmol/L (normally 20- 120 μmol/L) and 6.30±1.2 (normally 0.5-2.0), respectively. Moreover, level of phe and (phe/tyrosine) ratio after three hours of loading 100mg/kg body weight of phenylalanine were significantly elevated (P<0.001) reaching to 810.8±88.3 μmol/L and 22.13±1.9 respectively, suggesting a HPA and PKU cases. On the other hand, analytical results for loading test of Kuvan on Phe level, and Phe/tyrosine ratio showed that all 13 patients were (BH4) Kuvanresponsiveness (atypical phenylketonuric) as summarized in Table 2. The level of phe reduced to 407.7±36.2 μmol/L, 302.0±33.9 μmol/L, 261.5±24.6 μmol/L and 258.4±31.5μmol/L at 4, 8, 12 and 24 hrs. of Kuvan treatment respectively compared to 810.8±88.3 μmol/L at baseline. In addition, the phe/tyrosine ratio reduced and retained to the normal values 6.18±1.7; 7.11±2.1, 6.47±1.1 and 6.08±1.3 at 4, 8, 12 and 24 h respectively, on Kuvan treatment compared to 22.13±1.9 at baseline (Figure 1).
Table 2. Screening of atypical PKU patients treated with 20mg/kg body weight of Kuvan| parameters | Fasting | Baseline (3h after 100mg/kg phe intake) | Time after Kuvan treatment | |||
| 4hr | 8hr | 12hr | 24hr | |||
| Phe conc. μmol/L mean ±SD | 227.8±51.1 | 810.8±88.3** | 407.7±36.2* | 302.0±33.9* | 261.5±24.6 | 258.4±31.5 |
| Phe/tyrosine ratioMean ±SD | 6.30±1.2 | 22.13±1.9** | 6.18±1.7 | 7.11±2.1 | 6.47±1.1 | 6.08±1.3 |
Figure 1.Line chart of Phe/tyrosine ratio before and after Kuvan loaded test among studied patients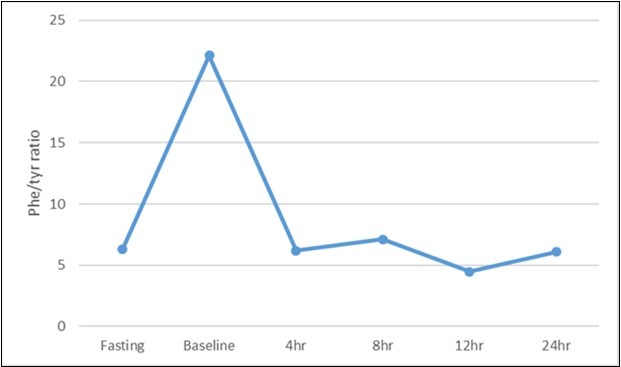
Mutation Analysis
Bi-directional Sanger sequencing of the PTPS gene on 13 unrelated Egyptian PKU-HB4 responsiveness patients’ and aligned with PTPS mRNA gene (GenBank NM_000317.3) revealed 6 different kinds of mutations with variable frequencies (one deletion mutation and 5 base substitution mutations; Table 3). Among studied patients, 61.5% (8/13) carried two variant alleles, either compound heterozygous (n = 2) or homozygous (n = 6) genotypes, all are common in same deletion mutation allele beside base substitution allele (three patients carried the same kinds of compound homozygous alleles). Other five patients (5/13; 38.5%) carried only one mutant homozygous allele; 4 of them carried the same deletion mutant allele, and one was base substitution mutation. The mutations were scattered across all six exons of the PTPS gene.
Table 3. Biochemical characteristics and PTPS mutations among atypical patients with PKU| patient | Nucleotide change | Exon. No | Amino acidchange | Consequence | State ofmutation |
| P1 | c.164_186del c.405T>C | del of all exon3 exon 6 | p.(Val55Aspfs*2)p.135Thr= | del Synonymous | Homozygous Heterozygous |
| P2 | c.164_186del | del of all exon3 | p.(Val55Aspfs*2) | del | Homozygous |
| P3 | c.164_186del | del of all exon3 | p.(Val55Aspfs*2) | del | Homozygous |
| P4 | c.200C>T | exon 4 | p.(Thr67Met) | missense | Homozygous |
| P5 | c.164_186del | del of all exon3 | p.(Val55Aspfs*2) | del | Homozygous |
| P6 | c.164_186del c.200C>T | del of all exon3 exon 4 | p.(Val55Aspfs*2) p.(Thr67Met) | del missense | Homozygous Homozygous |
| P7 | c.164_186del c.86A>T | del of all exon3 exon 2 | p.(Val55Aspfs*2 p.(Lys29Ile) | del Missense | Homozygous Homozygous |
| P8 | c.164_186del c.200C>T | del of all exon3 exon 4 | p.(Val55Aspfs*2) p.(Thr67Met) | del missense | Homozygous Homozygous |
| P9 | c.164_186del c.86A>T | del of all exon3 exon 2 | p.(Val55Aspfs*2) p.(Lys29Ile) | del missense | Homozygous Homozygous |
| P10 | c.164_186del c.200C>T | del of all exon3 exon 4 | p.(Val55Aspfs*2) p.(Thr67Met) | del missense | Homozygous Homozygous |
| P11 | c.164_186del c.22C>T | del of all exon3 exon 1 | p.(Val55Aspfs*2) p.(Arg8Cys) | del missense | Homozygous Homozygous |
| P12 | c.164_186del | del of all exon3 | p.(Val55Aspfs*2) | del | Homozygous |
| P13 | c.164_186del c.273G>A | del of all exon3 exon 5 | p.(Val55Aspfs*2) 91Lys= | del Synonymous | Homozygous Heterozygous |
Four novel mutations were identified in the present study; two variations that altered the coding sequences (c.22C>T and c.86A>T, resulting in p.Arg8Cys and p.Lys29Ile, respectively) and one variation produced silent mutation c.273G>A resulting in p.Lys=, in addition to a 23 base pair region deletion (c.164_186delAGTTGTGGTGACAGTACATGCAT resulting in p.Val55Aspfs*2). These four novel mutations were not detected in control chromosomes. Two base substitution mutations previously identified, one missense mutation c.200C>T and other produced a silent mutation c.405T>C resulting in p.Thr67Met and p.135Thr= respectively.
The most frequent mutation among studied patents with PKU is a novel deletion mutation represents about 92.3% occurrence of all MPKU patients. Deletion of 23 base pair at position c.164_186; p.(Val55Aspfs*2) in exon 3 was found in 12 patients (all patients except number 4) that produced a truncated un-functional protein. Different base mutations and corresponding amino acid sequences aligned with wild type PTPS gene as well as 3D structure of protein sequence as represented in Figure 2.
Figure 2.Chromatogram of novel deletion mutation. The upper part of panel A is wild type sequence and the lower part is the truncated mutant PTPS gene. Panel B is the 3D protein structure of PTPS in wild type and mutant one in upper and lower respectively.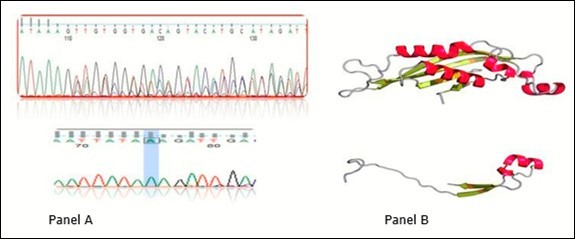
The second most frequent mutations among recorded PKU patients atypical type is previously published SNP mutation c.200C>T in exon 4 (Figure 3A) was found in patients number 4, 6, 8, 10, resulting in p.Trp67Met with a big change in 3D structure as represented in the solid line in yellow region (Upper part of Figure 3B). The degree of SNPs pathogenicity in exon 4 was determined by polyphen2 program, which represents a highly pathogenic mutation with score 0.995 where horizontal dark line in the red region (Lower part of Figure 3B).
Figure 3.Chromatogram of mutation of 200C>T on the lower part of panel A with wild type in lower part. Panel B represents the 3D structure in upper region and pathogenicity degree in the lower region A 200C>T mutation previously published. Polyphen2 data shows the mutation is probably damaging with score of 0.995 (sensitivity 0.99 specificity 0.99)
The third frequent mutation 86A>T in exon 2 is a novel mutation among patients representing p.Lys29Ile and located in patients 7 and 9 as represented in Figure 4A. The 3D structure and the degree of SNPs pathogenicity was determined by polyphen2 program, which represent a tolerated mutation due to represent of horizontal dark line in the green region (Figure 4B).
Figure 4.Chromatogram of novel mutations. Panel A sequence analysis of A>T 86 mutation in the lower part of panel A to compare with wild type sequence in the upper part. 3D structure of mutated protein in upper region and pathogenicity of the mutation by polyphen2. The probability is benign with score of 0.001 (sensitivity 0.99 and specificity 0.95)
The least frequent mutation represented only one patient of each mutation type. SNP 22C>T in exon 1 representing a novel missense mutation p.Arg8Cys in patient 11 (Figure 5A); while G>A at position 273 in exon 5 representing synonymous novel mutation p.91Lys= in patient 13 (Figure 5B) and a previously published 405T>C in exon 6 representing p.135Tyr= mutation in patient 1 (Figure 5c) are silent mutations. All are mild disease due to couple with deletion mutation.
Figure 5.Chromatogram of mutations represent the transition mutations where panel A shows 22C>T mutation (p.Arg8Cys), Panel B shows 273G>A and Panel C shows 405T>C. In the middle part is mutation compare to the wild type sequence in the upper part. 3D structures of mutation represented in the lower part of the panel. Last two mutations are silent mutations and 3D is similar. Ribbon diagram of a monomer of human PTPS depicting the locations of the amino acid replacements newly identified in current PTPS patients. Substituted amino acid residues denoted with yellow-colored ribbon
The pathogenicity degree of a novel missense mutation 22C>T; p.Arg8Cys was determined by polyphen2 program, which represent a tolerated mutation due to represent of horizontal dark line in the green region (Figure 6). Of course, other two synonymous mutations has no polyphen2 data.
Figure 6.Data of polyphen2 showed a cold pathogenicity of 22C>T mutation as represented by the vertical solid line in green region and cross to the left end, which is a benign mutation.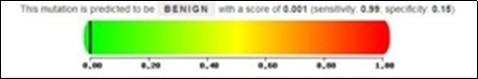
In current study, two software tools used to predict whether an amino acid substitution has an impact on the pathogenicity and biological function of a protein. PROVEAN and SIFT data for recorded mutations are listed in Table 4.
Table 4. The pathogenicity of identified mutations with two different predictor| Nucleotidechange | Amino acidchange | PredictionSIFT | PredictionPROVEAN |
| c.22C>T | p.(Arg8Cys) | deleterious | deleterious |
| c.86A>T | p.(Lys29Ile) | damaging | damaging |
| c.164_186del | p.(Val55Aspfs*2) | N/A | N/A |
| c.200C>T | p.(Thr67Met) | damaging | damaging |
| c.273G>A | 91Lys= | neutral | neutral |
| c.405T>C | p.135Thr= | neutral | neutral |
Discussion
Screening for PKU in newborns enables early diagnosis and therapeutic intervention to prevent the most severe consequences of disease disorder. The current standard therapy is adherence to appropriate treatment (including a phe-restricted diet) for life to maintain blood phe concentrations within recommended ranges to achieve the best clinical outcomes. While diet remains the cornerstone of treatment, in a subset of patients with absent or partial PAH deficiency activity, in another subset of PKU patients supplementation of tetrahydrobiopterin (BH4) alone or conjunction with a phe-restricted diet is essential to further lower elevated blood phe levels 28. Therefore, choice the way for HPA treatment is important since severe intellectual disability from PAH or BH4 deficiency is preventable with proper treatment.
Hyperphenylalaninemia (HPA) mainly caused by phenylalanine hydroxylase deficiency (PAHD; 98%) that called classical PKU disease or BH4 deficiency (2%; 13) that called atypical PKU disease. Internationally BH4 deficiency related mainly to PTPS deficiency, which is a major subset of cause atypical PKU (accounts for 65%), followed by quinoid dihydropteridine reductase (DHPR) deficiency (accounts for 25%; 29), and other three enzymes account for 10%. In the mainland of China, PTPS deficiency accounts for 96% and DHPR deficiency only accounts for 2.4% 30.
In 2014, the American College of Medical Genetics and Genomics (ACMG) published guidelines for treatment of PKU stipulated that BH4 responsiveness should be investigated by performing a BH4 loading test in all PKU patients 31 and European guidelines for the diagnosis and management of patients with PKU were published recently 32. In the present study, BH4 loading test carried out on all suspected PKU patients to select only 13 BH4 responsiveness patients for further molecular studying on PTPS gene for identification of different mutants located in this gene causing inability to synthesized BH4.
In the test, a baseline blood sample collected from food capillary of newborn patient or figure/vein of adults after 3 hr of introducing 100mg/Kg body weight phenylalanine dissolved in breast milk or water to measure phe level and phe/tyrosine ratio followed by administration of one dose 20 mg/kg Kuvan dissolved in breast milk or apple juice. Following Kuvan administration and throughout the entire test, a normal feeding regimen must be provided. Blood samples were collected to measure phe levels and phe/tyrosine ratio were taken at 4, 8, 12, and 24 h after Kuvan administration. Patients who will not respond to BH4, for longer than 24 h were excluded from our study. The results of 13 selected patients indicated a decrease in blood level of phe at any time point after loading of Kuvan of ≥30% compared with baseline indicates the presence of BH4 responsive HPA. Figure 7 represents the data of selected five patients with complete clinical, biochemical and unrepeated molecular data set. Muntau etal.,18 reported that, a rapid fall in the phe concentration to a value approaching the target range (120–360 μmol/L) within 4 h suggests a genetic disorder in the synthesis of BH4 known as primary BH4 deficiency caused mainly by PTPS or GCH1 deficiency. They also proved that if the blood phe concentration decreases to within the target range at 24 h, this suggested a DHPR deficiency or fully responsive PAH deficiency.
Figure 7.Illustrative of Kuvan loading test outcomes of phenylalanine concentrations at baseline, 4hr, 8hr, 12hr and 24hr among 5 atypical PKU studied patients corresponding to 5 different types of mutation in PTPS gene. Where mutations in p2 is only 23 base pair deletion, P6, P7, P11 and P13 are couple mutation 23 base pair deletion with previously identified 200C>T missense mutation; novel 86A>T missense mutation; novel 22C>T missense mutation; previously identified 273G>A missense mutation respectively.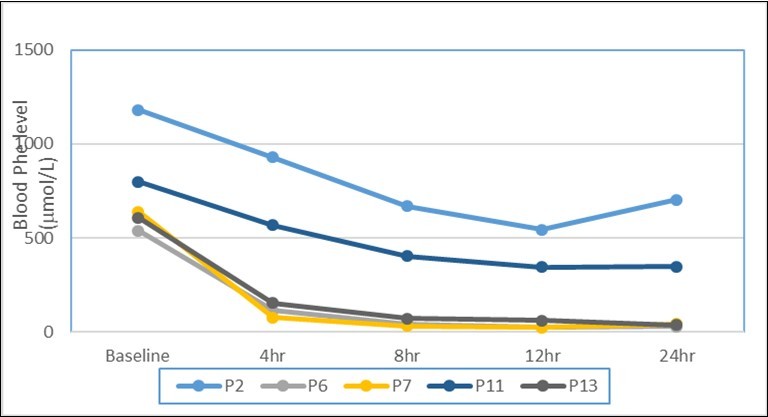
It is worth noting that the threshold of ≥30% is an arbitrary value; therefore, Muntau et al., 13 notice if the reduction in phe concentration is only slightly greater than 30% or is only observed at one test time point might not respond satisfactorily to sapropterin alone and must combined with a phe- restricted diet during a long-term therapy trial. Conversely, if the phe level dropped to more than 30%, (fully responsive patients) these patients showing a confident response to treatment with sapropterin alone. Hence, a positive BH4 loading test results should be critically reviewed in terms of phe tolerance. According to this assumption, all patients showed BH4 responsiveness and might be treated with only sapropterin with great consideration of patient number 1 who shows a reduction in phe level by about 23% only after 4hr of Kuvan treatment (Figure 7). Results of a BH4 loading test together with genotyping may help to estimate the probability of BH4 responsiveness and perfectly manage the treatment regime.
Therefore, it is important to detect BH4 responsiveness patients at the neonatal stage or even older to determine the appropriate course for managing the disease. In which only a subset of patients with PKU who are fully responsive to treatment with BH4, they will be treated only with sapropterin and those who are partially responsive will be treated with sapropterin and phe-restricted diet, and those who are not responsive will be treated with phe-restricted diet only.
Gene Mutation Analysis
In addition, sometimes BH4 loading test cannot distinguish patients with BH4 deficiency and BH4-responsive PKU 14. The conventional Sanger sequencing has been the gold standard test for a molecular approach in the genetic diagnosis of inherited PTPS disorders.
In humans, the PTPS gene (MIM 612719) located on chromosome 11q22.3-11q23.3, contains six exons and encodes for a protein of 145 amino acids 33. Reduction of the PTPS enzyme activity (MIM 261640) is the most common cause of patients with BH4 deficiency (approximately 59%; 34) causing hyperphenylalaninenia BH4-deficient type A, (HPABH4A).
Sanger sequence of 13 unrelated PKU patients identified 21 variants, 5 of them with a single mutation and 8 with two compound mutations representing a mutation detection rate of 100%. These mutations represented 6 different types of mutations (two of them are silent mutations, one novel c.273G>Al, p.91Lys= exon 5 and the other was previously identified c.405T>C, p.135Tyr= exon 6). Other four mutations represent three novel mutations, c.164_186del in exon 3 p.Val55Aspfs*2, c.22C>T, p.Arg8Cys in exon 1, and c.86A>T, p.Lys29Ile in exon 2, in addition to one previously identified c.200C>T, p.Trp67Met mutation in exon 4. Novel mutations were the most prevalent mutations in this study, which accounted for 76% (4/6) of the total mutant alleles. Particularly, p.Val55Aspfs*2 mutation has been reported as the most frequent mutation in PKU-BH4 responsiveness patients with a frequency of (12/13; 92%).
The most prevalent alleles, c.164_186del variant which accounted for 57% (12/21) of the total mutant alleles, c.200C>T, accounts for 19% (4/21), c.86A>T, accounts in 9.5% (2/21). The deletion mutations led to an in-frame deletion or frameshifting, while two missense mutations were predicted to affect enzymatic function and/or stability. However, to date, studies using exon analysis of different populations worldwide have shown that PTPS mutations were at least 88 different mutations 29, only two mutation in this study, missense c.200C>T, p.(Trp67Met) and silent mutation c.405T>C, p.135Tyr= were sited. Considering the relatively high number of patients with same deletion in our current population, postulate a founder effect of this mutation in the Egyptian population.
Since the incidence of PTPS deficiency is common among BH4 responsiveness patients, the distribution of common mutations were differ from population to population. In a previous study, PTPS deficiency was markedly higher (98%) in the Chinese population than that internationally accounted 29. The recorded results suggested that p.N52S, mutation in East-Asian populations and in Iranian patients 35, where p.V56M in western countries 36 and p.P87S in Chinese population have been reported as the most common mutations. This data explains that deletion mutation is the most common mutation among selected patients and very unique to them. Further studies on other Mediterranean area to confirm from this speculation. Although, novel deletion mutation is likely to be a pathogenic where, deletion mutations have been widely suggested to be disease-causing mutations 37. It should be highlighted that two novel missense variations c.22C>T, c.86A>T were observed in couple with frameshift mutation, there was supportive information that these new variations were likely to be pathogenic and may be disease-causing mutations. First, they also conserved to have same pattern of phe tolerance compared with deletion mutation only (Figure 7). Second, they are absent from the normal population and snpdb in National Center for Biotechnology Information (NCBI), similar to missense mutation c.200C>T previously identified as a pathogenic mutation in Italian patients 38. Third, a significant change in the chemical properties of amino acids in 3D structure (Figure 2, Figure 3, Figure 4, Figure 5). Forth, both mutations are predicted to be deleterious and damaging from prediction software SIFT and PROVEAN. To prove the function of these new variations will be further studied.
Molecular Modeling
On one other hand previously recognized c.200C>T; p.Trp67Met mutation 38 belongs to near region of catalytic domain (D89 and H 90 with C score 0.791 and 0.691 respectively, Figure 5). It was detected as a compound homozygous with del c.164_186; mutation in patients 4, 6, 8, 10 in the current study. Changing the Trp aromatic amino acid at position 67 to Met is expected to weaken the hydrophobic interactions of polypeptide coil region (from position 64 to position 69) among surrounding amino acids and the activity of PTPS may be dramatically decreased due to alterations in the side- chain sulfide bond that contribute to structural conformation. Figure 8.
Figure 8.Active sites of PTPS enzyme produced by I-TASSER software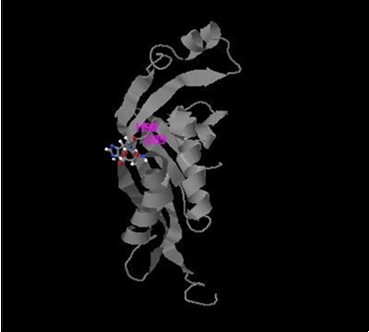
On the other hand, I-TASSER of two novel missense variants (p.Arg8Cys and p.Lys29Ile) identified in patient 11 and in patients 7&9 respectively as a compound homozygous with del c.164_186; mutation in PTPS protein are located away from the catalytic domain. Both mutations are located in the first and second coil region of PTPS polypeptide chain and predicted to be damaging and deleterious from prediction software, SIFT and PROVEAN (Table 4). Replacement of these basic amino acid Arg at position 8 and Lys at position 28 with neutral Cys and Ile amino acids respectively will change B-factor (is a value to indicate the extent of the inherent thermal mobility of residue in proteins in I-TASSER software). The mutation p.Lys29Ile is located very cross to ligand binding site residues for biopterins at position H24 and L26 with C-score 0.94, which considered more effective on PTPS function compared with p.Arg8Cys mutation in proximal region (Figure 9). This might explain the damaging effect of p.Lys29Ile compared with deterious effect of p.Arg8Cys mutations. Both novel missense mutations require more protein studying to analyze distortion of the backbone interactions among domains and changing in repulsive interactions.
Figure 9.I-TASSER results of PTPS wild type protein with legend and active sites.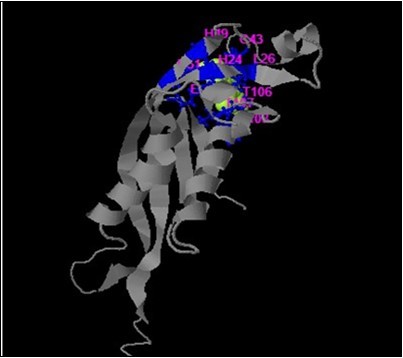
In summary, this is the first reported case of Egyptian BH4 deficient patients, which provides a reference for PTPS gene mutation research in the future. This study identified three different novel variations among 13 BH4- deficient patients to improve PTPS mutation database. The results could be of value for differential molecular diagnosis of BH4 deficiencies to avoid any delay in diagnosis and treatment.
Acknowledgments
The authors thank the family for their participation
References
- 1.G S Ribas, Sitta A, Wajner M, C R Vargas. (2011) Oxidative stress in phenylketonuria: what is the evidence?. , Cell. Mol.Neurobiol 31, 653-662.
- 2.R J Allen, Young W, Bonacci J, Persico S, Andruszewski K et al. (1990) Neonatal dystonic Parkinsonism, a "stiff baby syndrome" in biopterin deficiency with hyperprolactinemia detected by newborn screening for hyperphenylalaninmia and responsiveness to treatment. , Ann Neurol 28, 434-439.
- 3.P T Ozand. (1998) Hyperphenylalaninmia and defective metabolism of tetrahydrobiopterin. In: Nyhan WL, Ozand PT, (eds) Atlas of metabolic disease.Chapman & Hall Medical,London. 117-125.
- 4.Erlandsen H, A L Pey, A Pérez Gamez, Lourdes B, R. (2004) Correction of kinetic and stability defects by tetrahydrobiopterin in phenylketonuria patients with certain phenylalanine hydroxylase mutations. Proc. Natl Acad Sci USA 101 16903-16908.
- 5.Ozalp I, Coskun T, Tokatli A, H S Kalkanoglu, Dursun A. (2001) Newborn PKU screening in Turkey: at present and organization for future. , Turk J Pediatr 43, 97-101.
- 6.Aoki K, Ohwada M, Kitagawa T. (2007) Long-term follow-up study of patients with phenylketonuria detected by the newborn screening programme in Japan. , J Inherit Metab. Dis 30, 608-613.
- 7.Vockley J, Andersson H C, Antshel K M, Braverman N E, Burton B K. (2014) Phenylalanine hydroxylase deficiency: diagnosis and management guideline.Genet. Med.16: 188-200.
- 8.J G Loeber. (2007) Neonatal screening in Europe; the situation in. , J Inherit Metab Dis 30, 430-438.
- 9.Mei L, Song P, Xu L. (2013) Newborn screening and related policy against phenylketonuria in China. Intractable Rare Dis Res. 2, 72-76.
- 10.Pangkanon S, Ratrisawadi V, Charoensiriwatana W, Techasena W, Boonpuan K. (2003) Phenylketonuria detected by the neonatal screening program in Thailand. , Southeast Asian J Trop Med Public Health 34, 179-181.
- 11.Matalon R, Michals K. (1991) Phenylketonuria: screening, treatment and maternal PKU. , Clin Biochem 24, 337-342.
- 12.Targum S, Lang W. (2010) Neurobehavioral problems associated with phenylketonuria. , Psychiatry (Edgmont) 7, 29-32.
- 13.Yilmaz E, Cali F, Romano V. (2000) Molecular basis of mild hyperphenylalaninaemia in Turkey. , J Inherit Metab Dis 23, 523-527.
- 14.Blau N, J B Hennermann, Langenbeck U, Lichter-Konecki U. (2011) Diagnosis, classification, and genetics of phenylketonuria and tetrahydrobiopterin (BH4) deficiencies. , Mol Genet Metab 104, 2-9.
- 15.Pe y A L, Pérez B, L R Desviat, M A, Aguado Cristina et al. (2004) Mechanisms underlying responsiveness to tetrahydrobiopterin in mild phenylketonuria mutations. , Hum Mutat 24, 388-399.
- 16.C R Scriver. (2007) The PAH gene, phenylketonuria, and a paradigm shift. , Hum Mutat 28, 831-845.
- 17.A C Muntau, Roschinger W, Habich M, Demmelmair H, Hoffmann B. (2002) Tetrahydrobiopterin as an alternative treatment for mild phenylketonuria. , N Engl J Med 347, 2122-2132.
- 18.Muntau A, Moulin M, Feillet F. (2018) Diagnostic and therapeutic recommendations for the treatment of hyperphenylalaninemia in patients 0–4 years of age. , Orphanet Journal of Rare Diseases 1, 173-182.
- 19.Trujillano D, Perez B, Gonzalez J, Tornador C, Navarrete R. (2014) Accurate molecular diagnosis of phenylketonuria and tetrahydrobiopterin-deficient hyperphenylalaninemias using high-throughput targeted sequencing. , European Journal of Human Genetics 22, 528-534.
- 20.Keil S, Anjema K, Spronsen F J van, Lambruschini N, Burlina A. (2013) Long-term follow-up and outcome of phenylketonuria patients on sapropterin: a retrospective study. , Pediatrics 13, 881-1888.
- 21.Lia N, Yua P, Rao B, Deng Y, Guo Y. (2018) Molecular genetics of tetrahydrobiopterin deficiency in Chinese patients. , J Pediatr Endocrinol Metab 31, 1-6.
- 22.Thony B, Leimbacher W, Blau N, Harvie A, C W Heizmann. (1994) Hyperphenylalaninemia due to defects in tetrahydrobiopterin metabolism: molecular characterization of mutations in 6-pyruvoyl- tetrahydropterin synthase. , Am. J. Hum. Genet 54, 782-792.
- 23.Thony B, Auerbach G, Blau N. (2000) . , Tetrahydrobiopterin Biosynthesis, Regeneration and Functions Biochem J 1, 1-16.
- 24.D M BÜrgisser, Thony B, Redweik U, Hess D, C W Heizmann. (1995) 6-Pyruvoyl-tetrahydropterin synthase, an enzyme with a novel type of active site involving both zinc binding and an inter subunit catalytic triad motif; site-directed mutagenesis of the proposed active center, characterization of the metal bindingsite and modeling of substrate binding. , J. Mol. Biol 253, 358-369.
- 25.B A Stemerdink, A F Kalverboer, Meere J J van der, Molen M W van der, Huisman J. (2000) Behaviour and school achievement in patients with early and continuously treated phenylketonuria. , J Inherit Metab Dis 23, 548-562.
- 26.Blau N. (2013) Sapropterin dihydrochloride for the treatment of hyperphenylalaninemias. Expert opin drug metab toxicol. 9, 1207-1218.
- 27.Blau N, Yue W, Perez B. (2018) database on the Internet]. UK: Connecting rare disease researchers worldwide. C2006 -.
- 28.J, Wilcken B, Alexander I, Ellaway C, O'Grady H. (2005) Tetrahydrobiopterin-responsive phenylketonuria: the New South Wales experience. , Mol Genet Metab 86, 81-5.
- 29.Li L, Qin Y, Su Y, Jiang H, Rejiafu N. (2018) Gene mutation and pedigree analysis of tetrahydrobiopterin deficiency in a Uygur family of China. , J Clin Lab Anal 33, 1-6.
- 30.Ye J, Y L, W M Yu, Zou H, Jiang J. (2018) Demographics, diagnosis and treatment of 256 patients with tetrahydrobiopterin deficiency in mainland China: results of a retrospective, multicentre study. , J Inherit Metab Dis 36, 893-901.
- 31.Singh R, Rohr H, Frazier F, Cunningham D, Mofidi A et al. (2014) Recommendations for the nutrition management of phenylalanine hydroxylase deficiency. Gene. Med.16 121-131.
- 32.AMJ Van Wegberg, MacDonald A, Ahring K, Belanger-Quintana A, Blau N. (2017) The complete European guidelines on phenylketonuria: diagnosis and treatment.Orphanet. , J Rare Dis 12, 162-169.
- 33.Kluge C, Brecevic L, C W Heizmann, Blau N, Thony B. (1996) Chromosomal localization, genomic structure and characterization of the human gene and a retropseudogene for 6-pyruvoyl-tetrahydropterin synthase. , Eur J Biochem 240, 477-484.
- 35.Khatami S, S R Dehnabeh, Zeinali S, Thony B, Alaei M. (2017) Four years of diagnostic challenges with tetrahydrobiopterin deficiencies in Iranian patients. , JIMD Rep 32, 7-14.
- 36.Leuzzi V, C A, C L, Pozzessere S, Burlina A. (2010) Phenotypic variability, neurological outcome and genetics background of 6- pyruvoyl-tetrahydropterin synthase deficiency. , Clin Genet 77, 249-257.