Abstract
ALS is the neurodegenerative disease which is caused due to breakdown in interaction between UBL and rpn1. In this study, we explore the interaction of UBL and rpn1 which is involved in protein degradation. Protein recycling system plays a crucial role in degradation of deformed or damaged proteins. Task of degradation of damaged ubiquitinated proteins is completed by proteasome with the help of ubiquilin2 protein which links 19s proteasome and poly-Ub chain attached to damaged protein. More specifically, N-terminal UBL domain interacts with rpn1 subunit of base complex of 19s proteasome and C-terminal UBA domain interacts with tetra poly-Ub chain attached to damaged protein. In present study, UBL domains are docked against homology modeled rpn1 with the help of Patch dock server. Further the docked structures are refined using fire dock server and best docked structure is chosen having global energy -16.71. Best docked structures are analyzed using swiss-pdb viewer software to show hydrogen bonds between interacting proteins. Here we explore a mutation E6A and P11A in UBL structure with the help of YASARA which is significantly increasing the interaction between interacting proteins in terms of hydrogen bonds.
Author Contributions
Academic Editor: Tao Pang, Associate Professor, China Pharmaceutical University. Visiting Fellow, National Institute of Mental Health
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2014 N. Pradhan, et al
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Introduction
Amyotrophic lateral sclerosis (ALS), often referred to as "Lou Gehrig's Disease," is a progressive neurodegenerative disease that affects nerve cells in the brain and the spinal cord. Motor neurons reach from the brain to the spinal cord and from the spinal cord to the muscles throughout the body. The progressive degeneration of the motor neurons in ALS eventually leads to their death. When the motor neurons die, the ability of the brain to initiate and control muscle movement is lost. With voluntary muscle action progressively affected, patients in the later stages of the disease may become totally paralyzed.
Northwestern researchers have identified a link between ubiquilin-2 and the dominantly inherited chromosome X-linked Amyotrophic lateral sclerosis (ALS) and ALS/dementia. They discovered that ubiquilin 2, which is member of the ubiquilin family, regulates the degradation of ubiquitinated proteins. They show that mutations in UBQLN2 lead to an impairment of protein degradation. Ubiqulin 2 abnormalities are associated with abnormal protein aggregation and neurodegneration, revealing a common pathogenic mechanism that can be exploited for therapeutic intervention, including therapeutic screening and risk assessment of ALS and ALS-related diseases.
Ubiquilin-2 is a protein that in humans is encoded by the UBQLN2 gene 1. This gene encodes a ubiquitin-like protein (ubiquilin) that shares high degree of similarity with related products in yeast, rat and frog. Ubiquilins contain a "N-terminal ubiquitin-like domain and a C-terminal ubiquitin-associated domain. They physically associate with both proteasomes and ubiquitin ligases, and are thus thought to functionally link the ubiquitination machinery to the proteasome to effect in vivo protein degradation. In a small proportion of familial Amyotrophic lateral sclerosis (fALS), the UBQLN2 gene is mutated, causing formation of a non-functional Ubiquilin 2 enzyme. This non-functioning enzyme leads to the accumulation of ubiquinated proteins in the lower motor neurons and upper corticospinal motor neurons, due to the fact that ubiquilin 2 normally degrades these ubiquinated proteins, but cannot if the ALS mutation is present 2.
The Ubiquitin Proteasome Pathway (UPP) is the principal mechanism for protein catabolism in the mammalian cytosol and nucleus. Degradation of a protein via the Ubiquitin Proteasome Pathway (UPP) involves two discrete and successive steps: tagging of the substrate protein by the covalent attachment of multiple ubiquitin molecules (Conjugation); and the subsequent degradation of the tagged protein by the 26S proteasome, composed of the catalytic 20S core and the 19S regulator (Degradation) 3. This classical function of ubiquitin is associated with housekeeping functions, regulation of protein turnover and antigenipeptide generation.
Material and Methods
Retrieving Accession Number from NCBI-
Open NCBI and select the protein database. Search for UBL domain of ubiquilin2 or in place of ubiquilin2 write other synonyms of ubiquilin2 like Hplic2. Select the desired result i.e. 125 amino acid protein, solution structure of UBL domain of Hplic-2 and copy accession number.
Retrieving PDB Structure of UBL-
Crystal structure of Ubiquitin like domain of ubiquilin2 (Hplic2) from the organism Homo sapiens with the PDB ID 1J8C (Figure 1) is retrieved from the Protein Data Bank (PDB).
Figure 1. (PDB structure of UBL domain)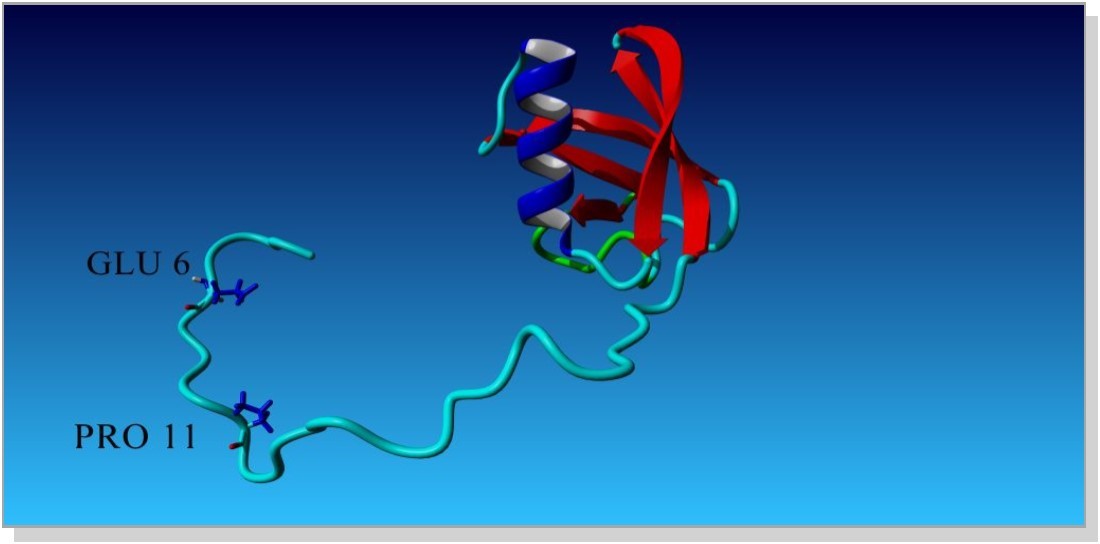
Homology Modeling-
Homology modeling, also known as comparative modeling of protein, refers to constructing an atomic-resolution model of the "target" protein from its amino acid sequence and an experimental three-dimensional structure of a related homologous protein (the "template"). Homology modeling relies on the identification of one or more known protein structures likely to resemble the structure of the query sequence, and on the production of an alignment that maps residues in the query sequence to residues in the template sequence. It has been shown that protein structures are more conserved than protein sequences amongst homologues, but sequences falling below a 20% sequence identity can have very different structure 4.
Modeler9.9 - MODELLER
software package (version 9v9) was used to construct ten homology models of the RPN1 protein using known structure of Crystal Structure Of S-Adenosyl-L-Methionine Methyl Transferase (Yp_165822.1) From Silicibacter Pomeroyi Dss-3 At 1.80 A Resolution having PDB ID 3IHT got after running blast. Structure having PDB ID 3IHT got after running blast was having 32% maximum identity.
Validation of 3-D Structure-
All predicted 10 models were evaluated by Procheck. Ramachandran plot statistics was used to evaluate the stability of model. Best structure was selected on the basis of maximum core region and dope score. 2nd model was chosen best having core region 79.5% and dope score -42516.06641. The resulting PDB structure of rpn1 subunit is shown in Figure 2 and corresponding Ramachandran plot is shown in Figure 3.
Figure 2. (Homology Modeled structure of RPN1 subunit)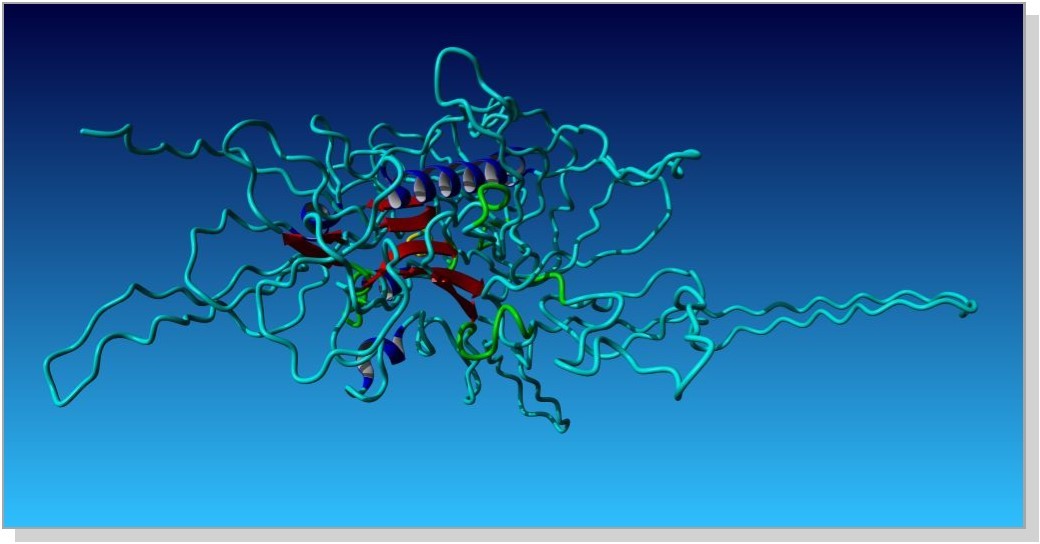
Figure 3. (Ramanchandran plot for 2nd Model having maximum core region)
Molecular Docking-
Binding of a small molecule (ligand) with a large molecule (protein) is called docking. Docking is the process by which two molecules fit together in 3D space. The objective of computational docking is to determine how two molecules will interact which will aid the interaction studies in bio-molecules. Molecular docking is often employed to aid in determining how a particular drug lead will interact to form a binding pocket.
Patchdock –
Patchdock algorithm 5 is inspired by object recognition and image segmentation techniques that are used in computer vision. Given two molecules, their surfaces are divided into patches according to the surface shape. All possible patches concave, convex or flat surface patches which can be visually seen are detected using segmentation algorithm. The patches are then filtered, so that only patches with hot spot residues are retained. Once the patches are identified, they are superimposed using shape matching algorithm. Shape matching algorithm uses hybrid of the geometric hashing 6 and pose clustering matching techniques to match the patches detected by segmentation algorithm. Concave patches are matched with convex patches and flat patches with any type of patches to obtain complexes. All the candidate possible complexes are examined.
Receptor UBL and ligand rpn1 molecule is uploaded in PDB format in Patchdock server, an automatic server for molecular docking. Clustering RMSD is chosen as 4.0 Å. E-mail address to retrieve the result is given. Complex type is chosen as default type. Then the docking job is submitted to the Patchdock server. Same steps are repeated for the mutated UBL and rpn1. Another docking job is submitted to the Patchdock server. Results are obtained through the e-mail address provided.
Firedock-
FireDock is an efficient method for refinement and re-scoring of rigid-body protein-protein docking solutions. Results from patch dock are refined by Firedock giving result ranked on the basis of global energy.
SPDBV-
Swiss-Pdb viewer (aka Deep View) is an application that provides a user friendly interface allow analyzing several proteins at the same time. It is used here for finding intermolecular hydrogen bond interaction between interacting proteins. Best docked structure from firedock result (Figure 9 and Figure 10) is selected on the basis of global energy and loaded in SPDB viewer. Hydrogen bonds are computed in structure and intermolecular hydrogen bonds are highlighted and noted. Same procedure is done for analyzing mutated docked structure.
Yasara-
YASARA is powered by PVL (Portable Vector Language), a new development framework that provides performance way above traditional software . PVL allows you to visualize even the largest proteins and enables true interactive real-time simulations with highly accurate force fields on standard PCs. You can push and pull molecules around and work with dynamic models instead of static pictures.
Here YASARA is used for replacing amino acid or setting mutation in UBL structure and for getting pdb structure of mutated UBL structure.
Results
The study involves the interaction of UBL and Rpn1.PDB structure of UBL domain is taken from NCBI as shown in Figure 1. The best homology model from ten models generated by Modeller9.9 is selected on the basis of Ramachandran statistics. PDB structure of modeled RPN1 subunit is shown in Figure 2 and corresponding Ramachandran plot for best model is shown in Figure 3. Mutated UBL (Figure 4) is docked against rpn1 subunit. The output of Patch Dock is a list of candidate complexes between UBL and rpn1 shown in Figure 5 and between mutated UBL and rpn1 is shown in Figure 6. The list is presented in the format of a table. Each table line represents one candidate complex. Solution No. represents the number of the solution. Score correspond to geometric shape complementarity score 4. The solutions are sorted according to this score. Area stands for the approximate interface area of the complex. ACE indicates Atomic Contact Energy 7. Transformations represented are 3D transformations that include 3 rotational angles and 3 translational parameters. These transformations are applied on the ligand molecule. PDB file of the complex denotes the predicted complex structure in PDB format. The corresponding fire dock solutions are shown in Figure 7 and Figure 8 respectively.
Figure 4. (PDB structure for Mutated UBL domain)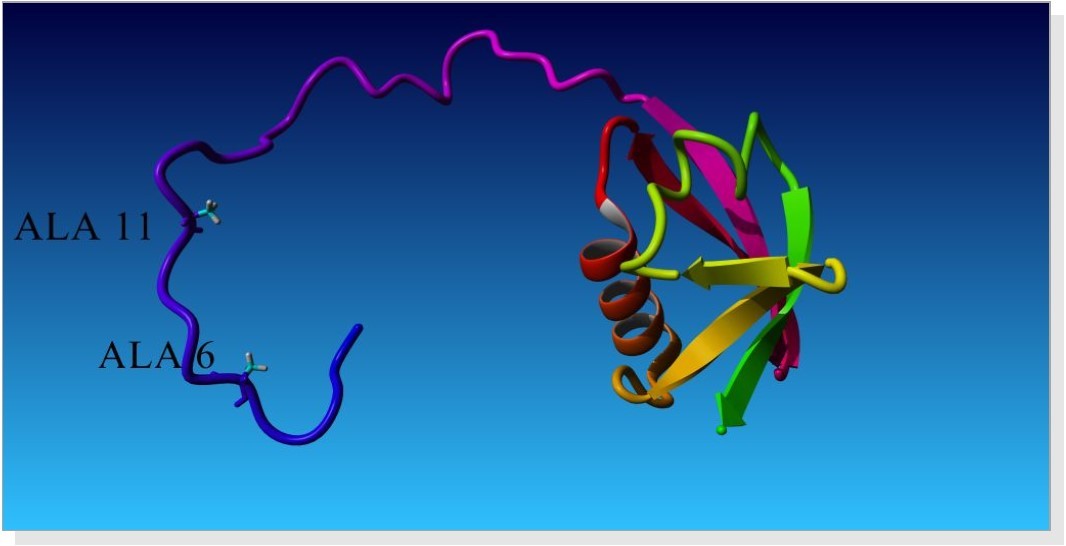
Figure 5. (Patch dock result for UBL and rpn1)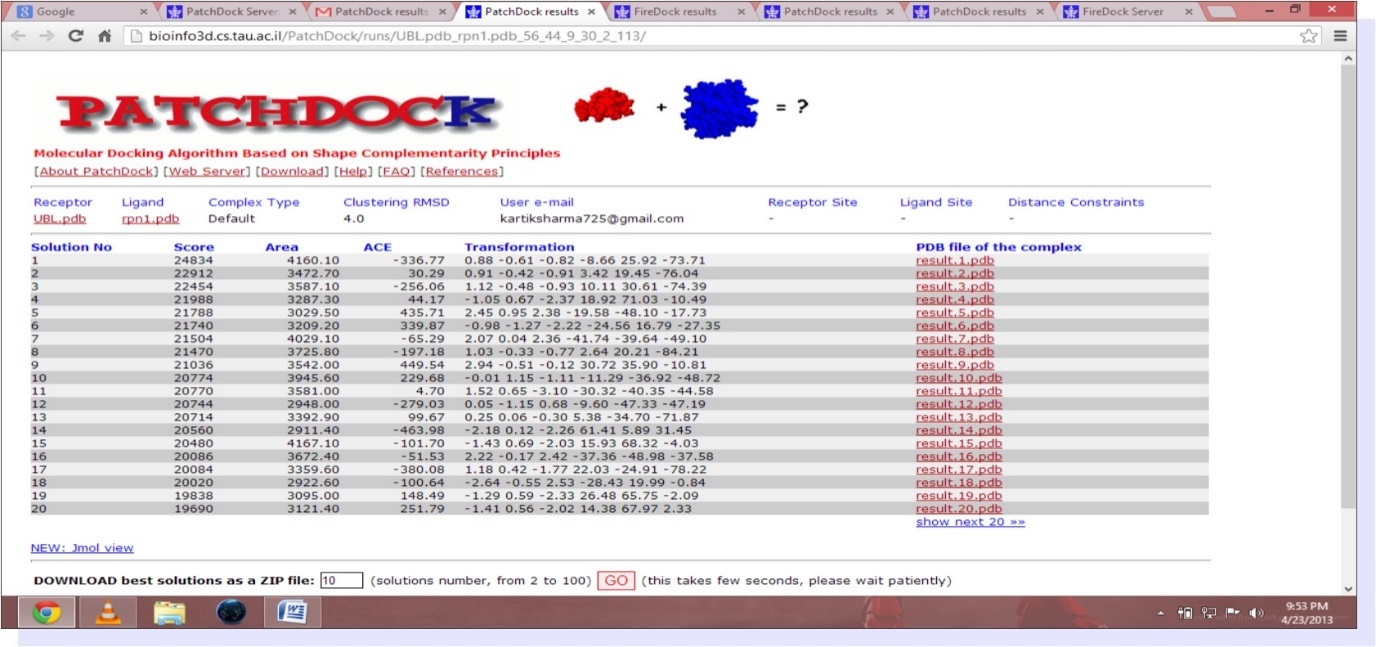
Figure 6. (Patch dock Result for mutated UBL and rpn1)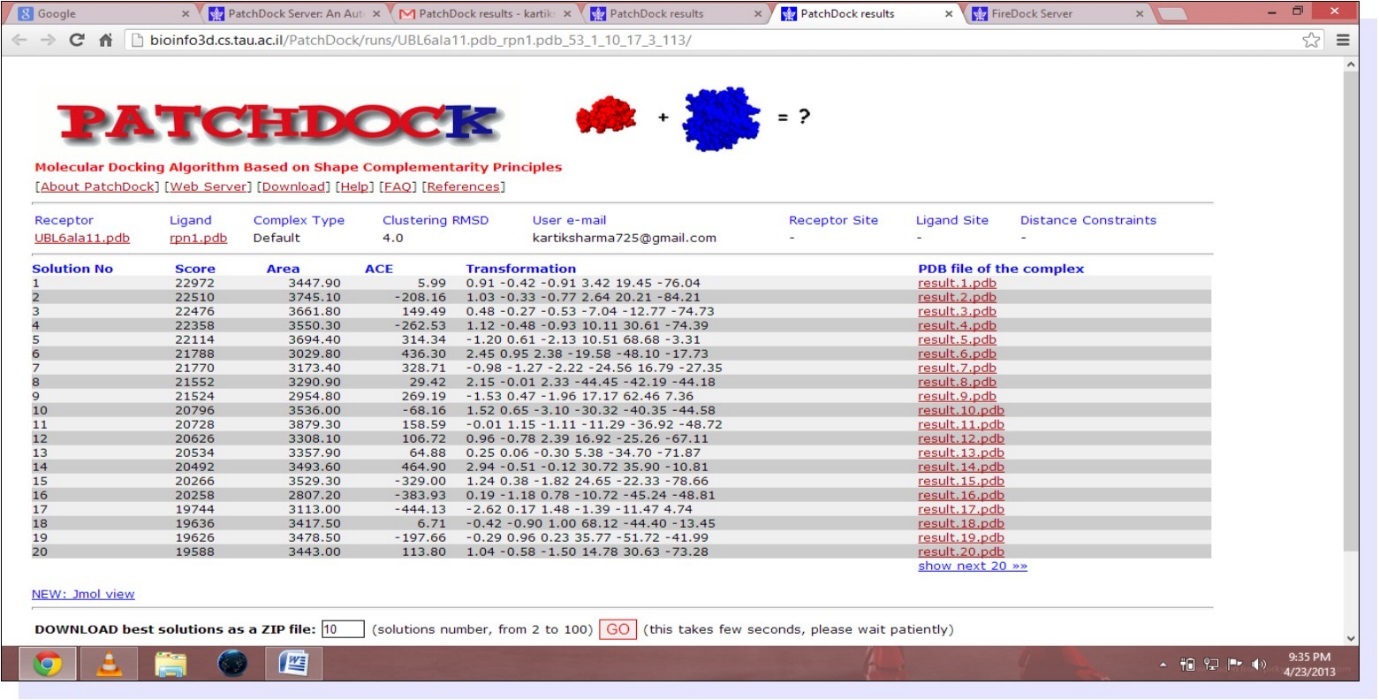
Figure 7. (Fire dock solution for UBL and rpn1)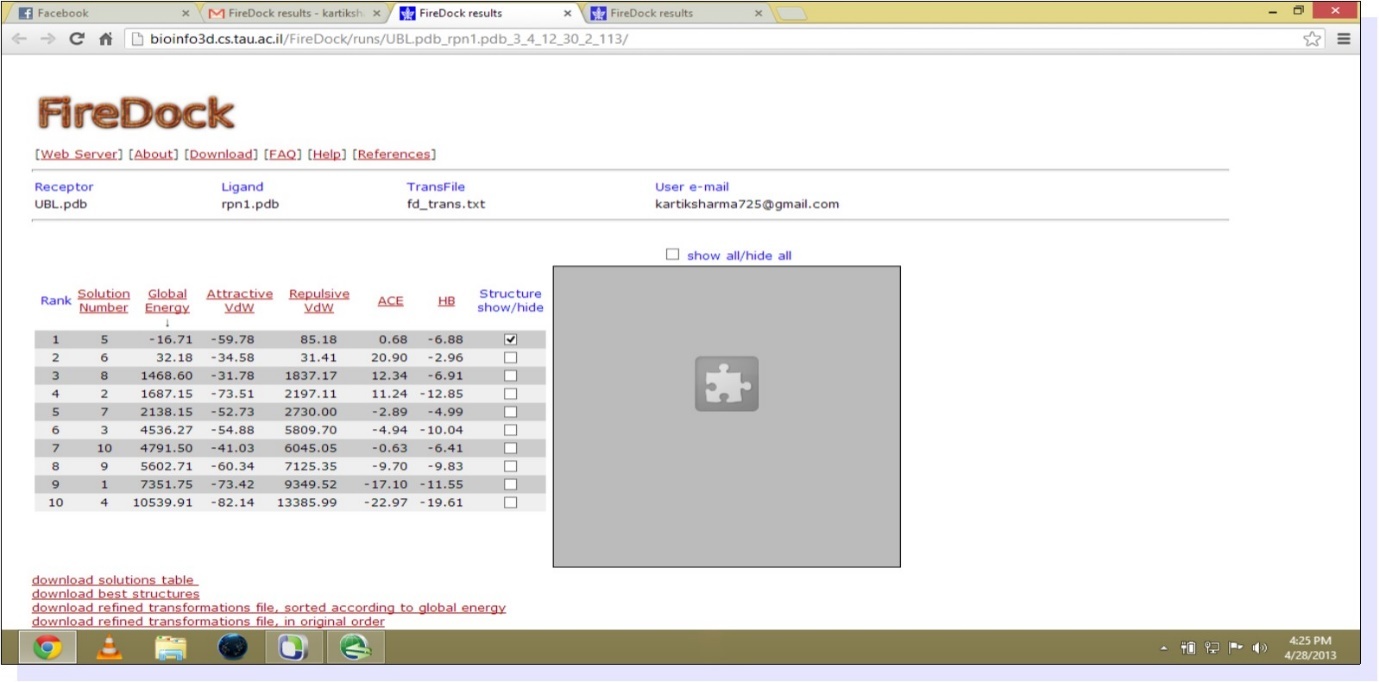
Figure 8. (Fire dock solution for Mutated UBL and rpn1)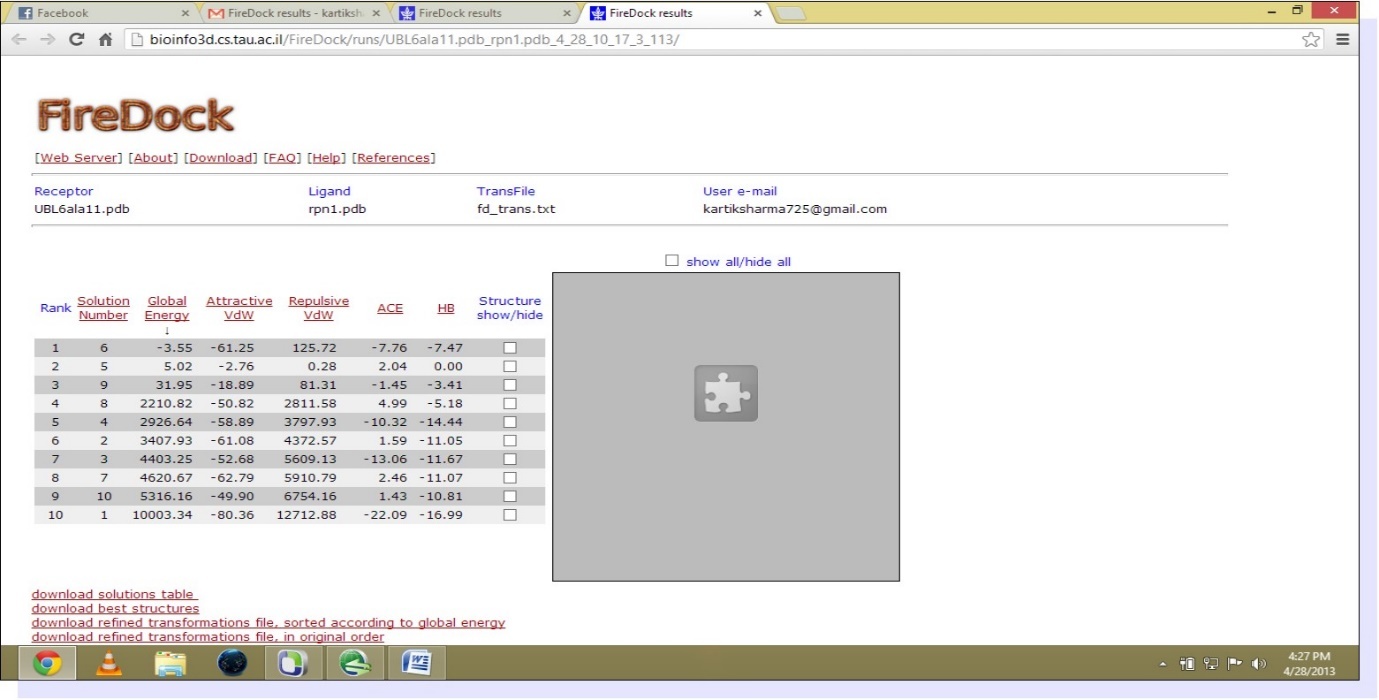
The hydrogen bond interaction between UBL and rpn1 are shown in Table 1whereas increased intermolecular hydrogen bond interaction after mutation in UBL are shown in Table 2and best docked structure refined by firedock for both cases are shown in Figure 9 and Figure 10 respectively.
Figure 9. (Best docked structure of UBL (Red) and rpn1 according to firedock results)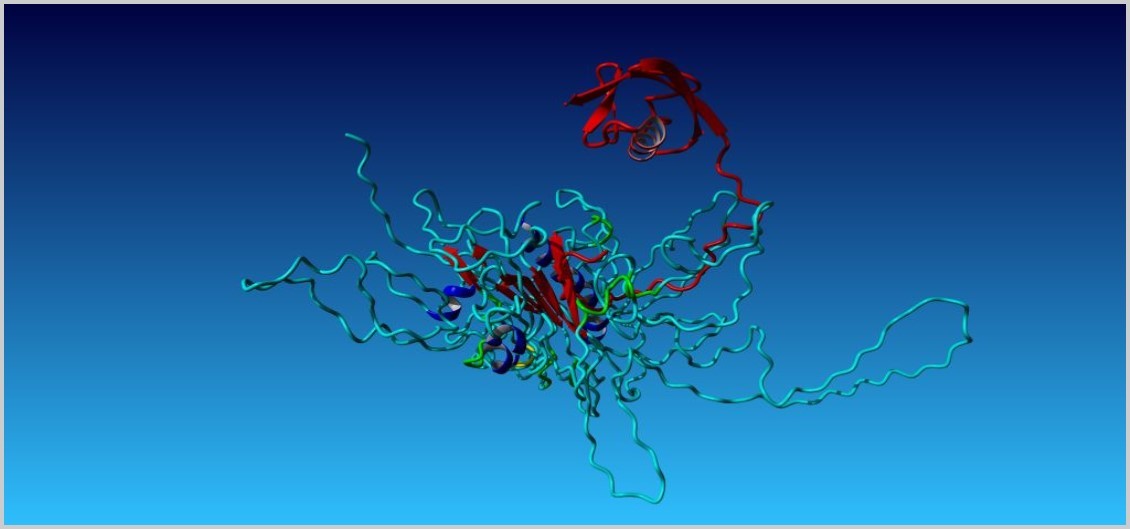
Figure 10. (Best docked structure of Mutated UBL (Red) and rpn1 according to firedock results)
Discussion
Degradation of misfolded, damaged, or otherwise malfunctioning proteins is certainly of utmost significance for cellular homeostasis and among the most important functions of the Ubiquitin-Proteasome System (UPS) .UPS includes interaction of UBL and UBA domains of ubiquilin2 gene with tetra-ub chain attached to damaged protein and rpn1, a non-ATPase subunit of 19s base complex respectively 8. In this study, we explore the interaction between two interacting proteins computationally and the result is explained in terms of intermolecular hydrogen bonds.
Structures of UBL and homology modeled RPN1 are analyzed separately using software such as SPDBV and YASARA.UBL domain of ubiquilin2 protein has 1586 atoms with 11070.674 g/mol mass while RPN1 subunit has 4845 atoms with 63675.646 g/mol mass.
PDB structure of UBL (ubiquitin like domain) is docked with the best structure based on maximum core region (79.5%) in Ramachandran plot of homology modeled RPN1 subunit. Protein- protein docking is performed by PATCHDOCK, an online server for molecular docking. PATCHDOCK give more than 2,000 resulting structure based on shape complementarity principle with their score, area and ACE values. Further, the results from PATCH DOCK are refined by another server FIREDOCK, which give us the 10 best structures. Resulted structures are ranked on the basis of their global energy. The best docked structure is selected on the basis of global energy which is -16.71. Results of fire dock are depicted in Figure 7.
Best docked structure is analyzed with the help of SPDB viewer. All intermolecular hydrogen bond including back bone and side chain interaction are considered and are shown in Table 1.
Table 1. (Hydrogen bonds in normal interaction)| ATOMS OF UBL DOMAIN | ATOMS OF RPN1 SUBUNIT |
|---|---|
| ASN4 ND2 | PRO75 O |
| ASN4 OD1 | PRO 75 N |
| ASN4 OD1 | GLN 152 NE2 |
| ASN4 N | GLU 150 O |
| ALA2 O | PHE 153 N |
| MET1 O | ILE 277 N |
| ALA2 N | SER275 O |
| GLU6 OE2 | ILE277 N |
| MET1 N | SER276 OG |
| GLU6 O | VAL155 N |
| ARG12 N | ARG111 O |
| PRO11 N | GLY110 O |
These intermolecular h-bond interactions are mainly responsible for proper functioning of UPS (ubiquitin proteasome system). These interactions triggers damaged ubiquitinated protein into proteasome. After a protein has been ubiquitinated, it is recognized by the 19S regulatory particle in an ATP-dependent binding step. The substrate protein must then enter the interior of the 20S particle to come in contact with the proteolytic active sites. Because the 20S particle's central channel is narrow and gated by the N-terminal tails of the α ring subunits, the substrates must be at least partially unfolded before they enter the core. The passage of the unfolded substrate into the core is called translocation and necessarily occurs after deubiquitination 3.
The mechanism of proteolysis by the β subunits of the 20S core particle is through a threonine-dependent nucleophilic attack 9. This mechanism may depend on an associated water molecule for deprotonation of the reactive threonine hydroxyl. Degradation occurs within the central chamber formed by the association of the two β rings and normally does not release partially degraded products, instead reducing the substrate to short polypeptides typically 7–9 residues long, though they can range from 4 to 25 residues depending on the organism and substrate.
In further study, analysis of UBL domain is done again by SPDBV and YASARA software. As ALS (amyotrophic lateral sclerosis), a motor neuron disease is caused by breakdown in protein recycling system or ubiquitin-proteasome system. It concludes there must be a mutation which is causing breakdown or significant decrease in number of intermolecular interaction in case of ALS.
YASARA, an interactive real-time molecular dynamics program is used for placing mutation in UBL structure and getting pdb structure of mutated UBL domain. We analyzed many mutations and finally we place a mutation E6A and P11A in UBL which results in significant increase in intermolecular hydrogen bond interaction.
Mutated UBL is having 1578 atoms and 10988.616 g/mol mass. There is a decrement in number of atoms and mass due to mutation as alanine is smaller than both proline and glutamic acid.
Mutated UBL and rpn1 are docked with the help of patch dock resulting in more than 1000 docked structure and these docked structures are refined by fire dock, online server for refining patch dock results. Ten best structures are sent to our e-mail ranked respectively on the basis of global energy. Best docked structure is chosen on the basis of their global energy which is -3.55(this value is considered to be related to free binding energy and higher negative value means higher free binding energy). Result after fire dock of mutated UBL and rpn1 is shown in Figure 8.Similar to first case, now best docked structure of mutated UBL and rpn1 are investigated with the help of SPDB viewer. All intermolecular hydrogen bonds are selected and revealed in Table 2.
Table 2. (Hydrogen bonds between mutated UBL and rpn1)| ATOMS OF UBL DOMAIN | ATOMS OF RPN1 SUBUNIT |
|---|---|
| ASN4 ND2 | ASN415 OD1 |
| ASN4 N | ASN415 OD1 |
| ALA2 N | GLU418 OE1 |
| MET1 O | THR499 N |
| SER7 OG | ARG494 N |
| SER7 OG | LEU492 O |
| ALA11 O | ARG507 NH1 |
| ARG12 NH1 | TYR503 OH |
| PRO13 O | ARG65 NH2 |
| SER14 O | TYR428 N |
| PRO17 O | ASN431 ND2 |
| ALA18 N | LYS432 O |
| ARG15 NH2 | GLU388 OE1 |
| ARG15 NE | GLU388 OE1 |
| ARG15 NE | MET435 O |
| ALA28 N | ARG536 O |
| ASN51 ND2 | SER43 O |
| ASN51 O | ALA47 N |
| PHE57 O | LYS564 N |
| SER52 OG | SER44 N |
| GLN56 NE2 | ILE589 O |
| SER52 N | GLU132 OE2 |
| SER52 N | HIS137 ND1 |
| PRO49 N | THR39 O |
| ALA47 O | ASP41 N |
| ALA47 O | ARG38 N |
| LEU87 N | GLY110 O |
| THR86N | GLY110 O |
| ASP85 OD1 | ARG278 NH1 |
| PHE46 O | THR39 OG1 |
| ILE32 N | LEU117 O |
| ALA77 N | ILE105 O |
| THR97 O | ILE105 N |
| ALA47 N | LEU10 O |
| ALA47 N | PHE9 O |
| VAL35 N | THR39 OG1 |
| LYS43 O | GLN552 NE2 |
| GLU45 OE2 | LEU13 N |
| LYS34 O | THR114 OG1 |
| LEU96 O | THR114 OG1 |
| LEU96 N | THR114 OG1 |
| LYS58 O | PRO337 N |
| THR97 OG1 | GLU93 OE2 |
| HIS99 ND1 | GLU93 N |
| HIS99 NE2 | GLN557 NE2 |
| HIS99 NE2 | GLU91 OE1 |
| ARG64 NH2 | LYS587 NZ |
| ARG64 NH2 | LEU577 O |
| GLU42 N | GLU560 OE1 |
| LYS38 O | LEU561 N |
| PHE65 O | TYR349 N |
| VAL101 O | LYS89 N |
| ILE75 N | VAL88 O |
| LEU74 O | SER109 N |
| LEU81 N | GLY107 O |
| VAL35 O | ARG38 NH2 |
| VAL35 O | ARG38 NE |
| ILE80 O | ARG111 N |
| LEU72 N | GLU584 OE1 |
| THR69 N | SER338 O |
| THR69 OG1 | ASN585 ND2 |
| ASP70 OD2 | THR403 OG1 |
| ASP83 OD1 | PHE156 N |
| GLN55 NE2 | ALA82 O |
| GLN56 NE2 | ILE589 O |
There was a significant increment in intermolecular hydrogen bond interaction. Twelve hydrogen bonds which were in normal case increased to sixty-five hydrogen bonds in mutated case and a global energy change from -16.71 to -3.55.This increment in intermolecular interaction can be seen as favorable in case of ALS. These increased interactions can lower the risk of ALS as increased interaction will reduce the risk of breakdown of protein recycling mechanism or interaction between UBL and rpn1.
In present study, it is indicated that certain mutation may cause enhanced interaction as shown in the results by mutation E6A and P11A in UBL domain. It enhanced the interaction between UBL and rpn1 by means of additional hydrogen bonds. Physiological significance of this study may be observed in future.