Abstract
In this theoretical discovery of a law of Life, there is MATHEMATICS (Geometry, Bits and Numbers) that UNIFY 3 universes as complementary as ATOMIC MASS, WAVES, and INFORMATION (DNA, RNA and Amino Acids). The discovery of a simple numerical formula for the projection of all the atomic mass of life-sustaining CONHSP bioatoms leads to the emergence of a set of Nested CODES unifying all the biological, genetic and genomic components by unifying them from bioatoms up to 'to whole genomes. In particular, we demonstrate the existence of a digital meta-code common to the three languages of biology that are RNA, DNA and amino acid sequences. Through this meta-code, genomic and proteomic images appear almost analogous and correlated. The analysis of the textures of these images then reveals a binary code as well as an undulatory code whose analysis on the human genome makes it possible to predict the alternating bands constituting the cariotypes of the chromosomes. The application of these codes to perspectives in astrobiology, cancer, and specifically in INFORMATION THEORY with the emergence of binary codes and regions of local stability (voting process), whose fractal nature we demonstrate, is illustrated.
PREFACE by Professor Luc Montagnier
Addendum by Robert Friedman M.D
After the discovery of the DNA double helix structure allowing both the stable storage of genetic information and its transfer through messenger RNA to protein synthesis organelles themselves structured by RNA most abundant in cells, the ribosomal.
This wonder of nature exists in ALL living beings from the virus to humans and is based on two codes, the linear sequence of nucleotides and that derived from codons where three nucleotides allow with a certain flexibility - synonymous codons - the choice in the twenty amino acids.
But we are missing a third CODE the one governing at multicellular beings from the rotifer to human, the stabilized modulation of gene expression in a nutshell the differentiation of cells from the single cell of the fertilized egg.
It is logical to think that this program which begins as soon as fertilization is written in the DNA.
We are also prone to associate it with non-coding DNA sequences although they control gene expression.
I introduce here the notion developed by Jean-Claude Pérez of mathematical harmony, a higher order present in all living beings and whose existence it finds in genomes, including those of viruses.
Thus the natural evolution of variants of the genome of coronavirus Covid 19 tends towards increasingly long Fibonacci series.
It remains to determine the Who, the How and the Why of such developments.
I will bet with my mathematician colleague that waves and fractals play a role.
Luc Montagnier
ADDENDUM
Jean-claude has given scientists a strong new direction for research. He has identified a unified field of science guided by the Golden Ratio and Fibonacci Sequence. By identifying an overall guiding principle that makes possible fractal-like nesting at all levels of biological manifestation, future researchers can begin with the "whole" instead of the "parts". If we know that complex systems are organized at varying levels by the Golden Ratio and Fibonacci Sequence, we can look for those universal patterns first and then fill in the gaps with small details to complete the picture. It's like having an overall view of a crossword puzzle before beginning to assemble the individual pieces. Without an overarching vision and guiding principle, completing the puzzle is infinitely more difficult. Once scientists and researchers realize and begin using this "SECRET IN HIDDEN IN PLAIN SIGHT," their discoveries will be orders of magnitude more fruitful.
Robert Friedman M.D
Author Contributions
Academic Editor: Dumrul Gulen, Kemal University Institude of Healyh Sciences Tumor Biology & Immunology Department.
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2021 Jean-claude Perez
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Introduction
Beyond its appearance, the major subject of this article will not concern biology, genetics or genomics, but rather the question of the emergence of COMPLEXITY, in the line of research like those of Ilya Prigogine, Benoit Mandelbrot or John Nash 1, 2, 3.
Indeed, the central question will be that of the emergence of "regularites" from an apparent disorder. The best illustration is that of the emergence of Benard's cells 1, putting organization way from the balance of billions of atoms, or the Mandelbrot's question "How Long Is the Coast of Britain?” 2.
Thus we will demonstrate how a binary code can emerge from the apparent disorder of DNA at the level of the entire human genome. We will also show how the living processes constituting the biosphere organize the proportions of the different atomic masses mono-isotopic of the same atom (oxygen for example) then produce the "regularity" of the average atomic masses which prove to be optimal vis avis of the code of the “atomic masses code” discovered here (§Methods Atomic code -I-).
Twenty years ago, in 1997, it was a kind of "scientific aesthetic sense" that motivated the following research: we found it abnormal that life needed 3 languages to code the information of the living: DNA, RNA, and amino acids, language of proteins ... this luxury of redundancy seemed illogical to us, if only because of this famous maxim "All that is simple is false, all that does not is not is unusable ". Others will say "small is beautiful" ... We could add "all that is simple is beautiful" ... It is true that from 1990 we demonstrated how the Fibonacci numbers - key esthetic of the nautilus, the pineapple or pine cones - structured the DNA sequences of genes (4, 5) or small genomes such as mitochondrial DNA 6.
We were looking for a smaller common denominator for DNA, RNA, and amino acids; we have the intuition of the need of 3 "ingredients":-the atomic mass and the bioatoms C O N H S P are common to these 3 languages.-the 2 universal constants PI and Phi could play a role.
Finally, we imagine a kind of digital "projection" (such as those of the cosine or the sinus in geometry), projection which would constitute a kind of "shadow" projecting on the 2D horizontal plane the image of a kind of complex "meta-code" common to these 3 languages.
Then we discovered then publish 7, 8 six “russian dolls like” embedded CODES
-I-Atomic mass code.
-II-Master code.
-III-Binary code.
-IV-Undulatory code.
-V-Cytogenetic code.
-VI- Standing waves meta-code.
Methods
We describe here the 6 embedded steps of 'Fractal Life Codes”
-I-Atomic mass code.
-II-Master code.
-III-Binary code.
-IV-Undulatory code.
-V-Cytogenetic code.
-VI-²Standing waves meta-code.
Their nature is fractal, each new code is based on entries on the previous code (s).
-I-Atomic Mass Code
Function
Transform any atomic mass (real number) into an integer number between -3 and +7, corresponding to multiples of Pi / 10 (-3Pi / 10 ... / ... + 7Pi / 10).
Inputs
A real number corresponding to any atomic mass mono-isotopic, average, or composed of one or more atoms
Outputs
the « Pi-mass », an integer number between -3 and +7, corresponding to multiples of Pi / 10 (-3Pi / 10 ... / ... + 7Pi / 10).
Summary
A quick presentation of the formula for life: In 5, 6, 14, 15 we introduced the law we call Formula for Life. This law unifies all of the components of living including bio-atoms, CONHSP and their various isotopes, to genes, RNA, DNA, amino acids, chromosomes and whole genomes. This law is the result of a simple non-linear projection formula of the atomic masses. The result of this projection is then organized in a linear scale of integer number based codes (e.g., -2, -1, 0, 1, 2, 3...) coding multiples Pi/10 regular values. These codes are called Pi-masses.
Process
Computing the “Formula for Life” associated with any atomic mass of Life components:
For atomic mass of any biological compound, we operate the “projection” of the atomic mass numerical value using the following operator:
where 
then P = 0.742340663...
Now, consider the “v” value, where v is always a negative or zero real number.
Then consider the function:
Where Abs (v) is the absolute value of v, and « remainder » or « residue » the decimal remainder of the numerical projection
For example: remainder (-27.85) = 28-27.85 = 0.15
We then defined PPI (m) such that:
 Note that (1-P.PI) is always negative because m is always positive, and (1-P.PI) is always negative.
Note that (1-P.PI) is always negative because m is always positive, and (1-P.PI) is always negative.
As an example, consider the amino acid GLY:
We defined the average mass of GLY as: GLY = 75.067542
Then: (1-P.PI) . GLY = -99.99987286
Thus, PPI (GLY) = remainder (1 = 0.0001271351803
Then finally, the result is a real number which we retain only the residues (decimal remainder),
PPI (GLY) = 0.0001271351803
Although no longer considered the decimal part, we note that, if we were interested in the set
(1-P.PI) . GLY = -99.99987286, this value is substantially equal to 100 = 10*2 ... which is not “just any number” ... So then, what is the geometric reality of this projection? As Figure 1, Figure 2 summarizes above, everything happens as if the atomic mass was “filtered” through the competitive interference of two projections: one through a cube of side = 1 and the second through of a sphere of radius = φ × 7/4.

Figure 2. Geometric meaning of the formula for life numerical projection
More precisely, let's take an example, by extending the example already presented relating to Glycine (GLY):
We calculated PPI (GLY) = 0.0001271351803.
We can then calculate the 21 PPI (GLY) -R (N.PI / 10) deviations where R (N.PI / 10) is the rightmost column in the previous table of N.PI / 10.
The following 21 values are then obtained:
0.8582802112 0.1724394766 0.4865987419 0.8007580073 0.1149172727 0.429076538 0.7432358034
0.05739506874 0.3715543341 0.6857135995 0.0001271351803 0.3140321302 0.6281913955 0.9423506609
0.2565099263 0.5706691916 0.884828457 0.1989877223 0.5131469877 0.8273062531 0.1414655184
It can be seen that the minimum difference (underlined) corresponds to an angle of 0 °, so to N = 0.
So we'll say that "PI-MASS (GLY) = 0"
The successive stages characteristic having transformed the mass GLY in PI-MASS = 0 are:
Atomic mass GLY = 75.067542
PPI projection (GLY) = 0.0001271351803
Angle N.PI / 10 the nearest: 0.000000000 (N = 0)
Approximate error: EPI (GLY, 0) = 0.0001271351803
PI-MASS (GLY) = 0 either 0 ° or else 0.PI / 10
As a documentary, we will calculate the PI-MASSES relating to 10 sgnificant different genetic materials. Consider any atomic mass « m », which may be that of a bio-atom, of a nucleotide, a codon or an amino acid or any other genetic compound based on bio-atoms or even, any atoms from Mendeleiëv periodic table.
This process will work especially on the average masses (mix of various isotopes % proportions). But it may also be applied to a particular isotope or any derivative of specific atomic mass proportions of the various isotopes.
A startling observation opens the door to enormous opportunities in astrobiology: Table 4 and Figure 3 shows a very curious fact: the Pi-mass projection formula seems optimal only for the atomic masses of average atomic weights of basic life bioatoms C O N H. Instead of tiny perturbations on these atomic masses and atomic masses of the individual isotopes (example O16) of each of these atoms “destroy” the optimality and fine-tuning of these projections then, also, consequently all resulting master code perfect tuning. Example here (Table 4) for the Pi-mass projection of Oxygen isotopes and % average weighted atom mass. As shown in Figure 3, isotopes of oxygen lightest and heaviest O16 O18 both produce an error on the projections Pi-mass much higher than that of the average atomic mass of that atom of oxygen consisting of: 99,757% + 0.04% O16 O17 + 0.2% O18. Table 1, Table 2, Table 3, Table 4, Table 5
Table 1. A set of Pi-mass projections for some main Life compounds| Nature | Molecule or bioatom | Average atomic mass | Projection PPI(m) | Pi-mass NPI(m) | Angle | Error EPI(m,N) |
|---|---|---|---|---|---|---|
| #NAME? | ||||||
| Bioatom | C12 Carbon isotope 12 | 12 | 0.014416319 | 0 PI/10 (0°) | 0° | 0.014416319 |
| Bioatom | C (Carbon average mass) | 12.0111 | 0.000370346 | 0 PI/10 (0°) | 0° | 0.000370346 |
| Nucleotide | (G nucleotide) | (G nucleotide) | (G nucleotide) | 0 PI/10 (0°) | 0° | 0.019744693 |
| Codon | Codon TCA | 369.324471 | 0.011063612 | 0 PI/10 (0°) | 0° | 0.011063612 |
| Codon | Codon UCA | 355.297477 | 0.69687081 | -1 PI/10 (-18°) | -18° | 0.011030076 |
| Codon | Codon AGT (TCA complement) | 409.349065 | 0.693022221 | -1 PI/10 (-18°) | -18° | 0.007181486 |
| double-stranded DNA | DNA double strand : TCA+AGT | 778.673536 | 0.704085833 | -1 PI/10 (-18°) | -18° | 0.018245098 |
| Amino acid | PRO (Proline amino acid) | 115.13263 | 0.628142392 | +2 PI/10 (+36°) | +36° | 0.000176139 |
| Amino acid | LYS (Lysine amino acid) | 146.190212 | 0.255344393 | +4 PI/10 (+72°) | +72° | 0.001292669 |
| Peptide link | CONH Peptidic link | 43.025224 | 0.684723446 | -1 PI/10 (-18°) | -18° | 0.001117289 |
| -3 PI/10and less | -2 PI/10 | -1 PI/10 | 0 PI/10 | +1 PI/10 | +2 PI/10 | +3 PI/10 | +4 PI/10 | +5 and +7 PI/10 | |
| Bioatoms | P(-4pi/10) | H O | C | N | S | ||||
| Nucleotides | U G I | T C A | |||||||
| Others compounds | Ph/sugarRNA | CONH | H2O | CH2Ph/sugar DNA | |||||
| Amino acids | Asp | Asn GluGly Ser | Ala GlnHis Thr | Pro Tyr Cys (+2) | Arg PheTrp Val | Ile Leu LysMet (+4) | Cys (+5)Met (+7) | ||
| Codons DNA | ggg | gtg gcg gag tggcgg aggggt ggcgga | ttg ctgatg gttgtc gtatcg ccgacg gctgcc gcatag cagaag gatgac gaatgt tgctga cgtcgc cgaagt agcaga | ttt ttctta cttctc ctaatt atcata tcttcc tcacct ccccca actacc acatat tactaa catcac caaaat aacaaa | |||||
| Codons RNA | uuu uugguu gugugu uggggu ggg | uuc uuacuu cugauu augguc guaucu ucggcu gcguau uaggau gagugc ugacgu cggagu aggggc gga | cuc cuaauc auaucc ucaccu ccgacu acggcc gcauac uaacau cagaau aaggac gaacgc cgaagc aga | ccc ccaacc acacac caaaac aaa |
| Genetic compounds | Atomic mass m | PI-mass N PI/10 | Error EPI(m,N) |
| Regular GLYCINE Amino acid.GLY=NH2-CH2-COOHHYDROGEN atom mass H=1.007947 | 75.067542 | 0 PI/10 (0°) | 0.0001271351803 |
| GLYCINE modified by the atomic mass of only one of the HYDROGEN atoms that becomes H*=1.0080424374(the other H remain unchanged).GLY=NH2-CHH*-COOH | 75.06763744 | 0 PI/10 (0°) | 3.173283858 10*-11soit 0.0000000000317 |
| Electron (à titre indicatif) | 0.000549 | 0 PI/10 (0°) | 0.0007313405 |
| Atom | Isotope | Relative atomic mass | % isotopic composition | Pi projection residue and Pi mass value | Pi-mass NPI(m)=N.Pi/10 | Error EPI(m,N) |
| Oxygen | Average % balance | 15.9994(3) | - | 0.686647751 0.685840735 | -1 | 0.000807016 |
| O16 | 15.994 914 619 56(16) | 0.997 57(16) | 0.692662834 0.685840735 | -1 | 0.006822099 | |
| O17 | 16.999 131 70(12) | 0.000 38(1) | 0.354913152 0.371681469 | -2 | 0.016768318 | |
| O18 | 17.999 161 0(7) | 0.002 05(14) | 0.022742056 0.000000000 | 0 | 0.022742056 |
Figure 3. The OPTIMALITY of the average atomic mass is proved here on the PI-masses of ten primordial organic components.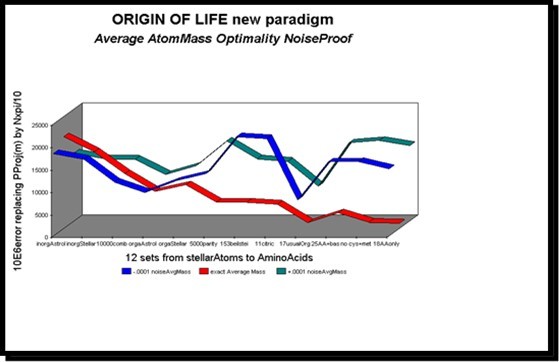
-II-Master Code
Function
Global integration of Genomics and Proteomics Pi-mass codes at the whole sequence level.
Inputs
DNA double stranded sequence by codons pairs producing Genomics (DNA) Pi-mass code and Proteomics (coresponding potential amino acid) Pi-mass code (an integer number for each codons pair)
Outputs
2 Genomic and Proteomic numerical vectors generating 2 patterned 2-D Genomic and Proteomic Images signatures.
Summary
Starting from the atomic masses constituting nucleotides and amino acids, a numerical scale of integers characterizing each bioatom, each TCAG DNA base, each UCAG RNA base, or each amino acid, an integer numbers scale code is obtained. Then, for each sequence of double stranded DNA to be analyzed, the sequence of integers that characterizes it (genomics) is constructed as well as the sequence of amino acids that would encode this double strand if each of the strands was a potential protein (proteomics). The remarkable fact is that this proteomics image still exists, even for regions not trans lated into proteins (junk dna). The computational methodology of the Master code (3, 4) then produces 2 patterned images (2D curves, see Figure 4, Figure 5, Figure 6) which are very strongly correlated. This would mean that beyond the visible sequence of DNA there would be a kind of MASTER CODE being manifested by two supports of biological information: the sequences of DNA and of amino acids, the RNA image constituting a kind of neutral element like the zero of the mathematics (Figure 4). Our thousands of genes and genomes Master Code analyses (viruses, archaeas, bacteria, eucharyotes) demonstrated that the extremums (max and min) signify functional regions like proteins active sites, fragility points like chromosomes breakpoints).
Figure 4. A symbolic representation of the 3 worlds of double stranded DNA (Genomics) highly corelated with potential double stranded amino acids (Proteomics) while RNA double stranded image is like a neutral element.
Figure 5. illustration of the high correlation coupling between genomics and proteomics images of a 100kb stretch of chromosome 7 (99.93% correlation).
Figure 6. illustration of the high correlation coupling between genomics and proteomics images of a complete MALARIA chromosome (98.08% correlation).
Figure 7. Evidence of first-order differential texture and roughness analysis (Leibniz) on Master Code Genomics and Proteomics images.
Figure 8. Clouds of points - In whole chromosome 22, population of Genomics curves will be relatively dispersed around one single withdrawing attractor in a kind of Gaussian dispersion (red), while the population of Proteomic curves will be distributed around two binary attractors (blue).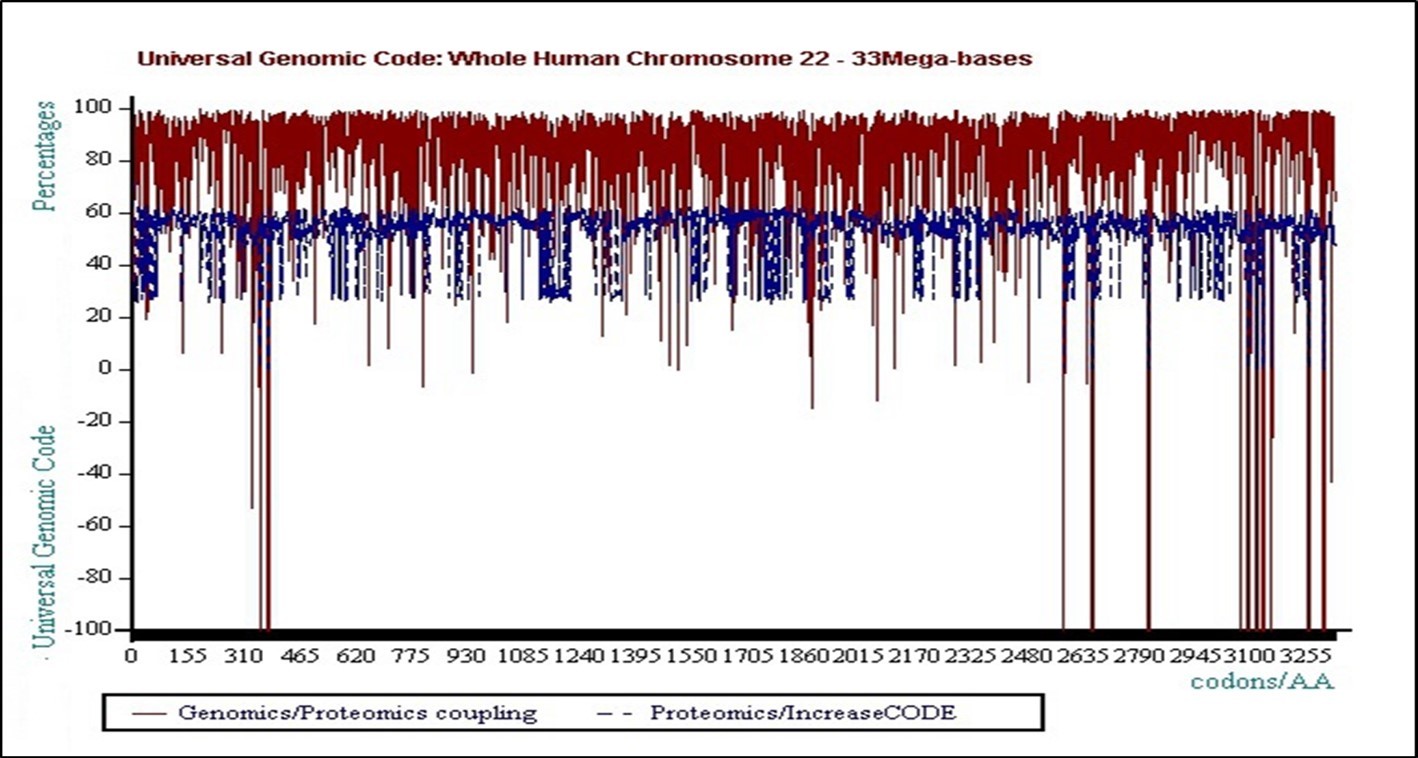
Figure 9. Frequency distribution - In whole chromosome 22, population of Genomics curves will be relatively dispersed around one single withdrawing attractor in a kind of Gaussian dispersion (red), while the population of Proteomic curves will be distributed around two binary attractors (blue)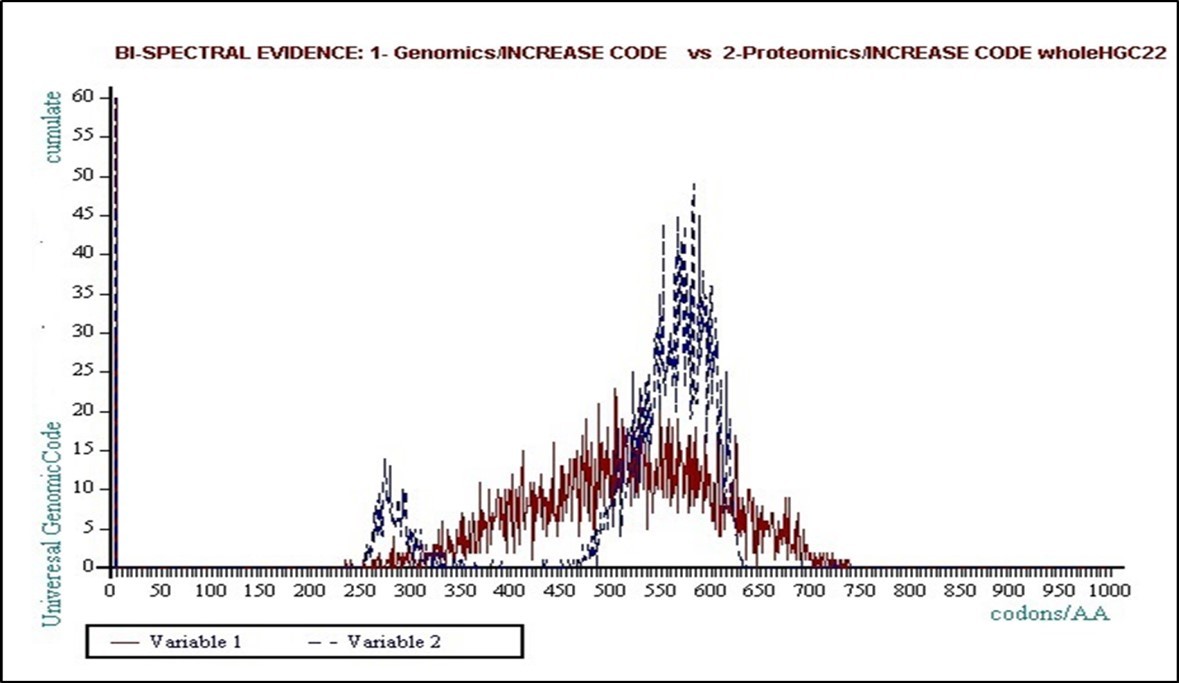
Figure 10. whole chromosome 4 – evidence of the perfect Proteomics Binary Code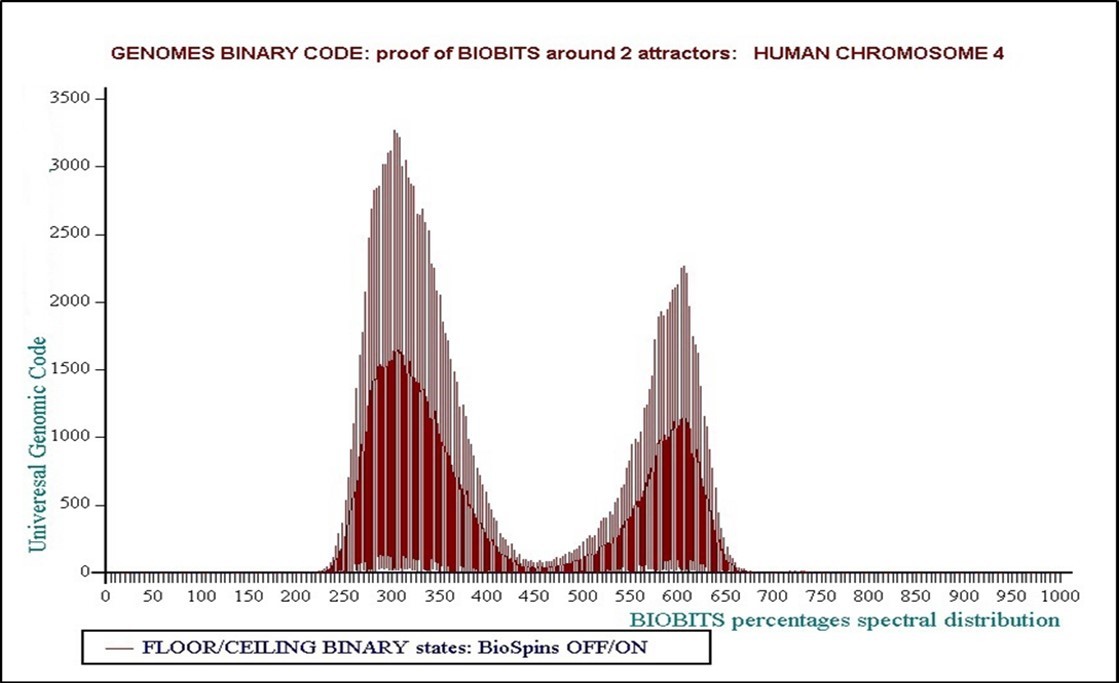
Process
An overviev on the “Biology Master Code” Great Unification of DNA, RNA and Amino acids : this process run 3 sequential substeps :
a) The coding step
b) The globalization and integration step
c) The great Unification between Genomics and Proteomics Master Code images
It may seem surprising that such a fine tuned process like biology of Life requires the use of three languages as diverse and heterogeneous as DNA with its alphabet of four bases TCAG; RNA with its alphabet of four bases UCAG; and proteins with their language of 20 amino acids. Obviously, the main discoveries in biology were made by those who managed to unearth the respective areas and “bridges” between these three languages. How ever, any “aesthete” researcher will think the table of the universal genetic code seems rather “ad hoc” and heterogeneous.
Starting only from the double-stranded DNA sequence data, the “Master Code” is a digital language unifying DNA, RNA and proteins that provide a common alphabet (Pi-mass scale) to the three fundamental languages of Genetics, Biology and Genomics.
The construction method of “the Master Code” will be now fully described below. It will highlight a significant discovery we summarize as follows: “Above the 3 languages of Biology - DNA, RNA and amino acids, there is a universal common code that unifies, connects and contains all these three languages”. We call this code the “Master Code of Biology.”
Here is a brief description of our process for computing the Master Code
a) The Coding Step
First, we apply it to any DNA sequence encoding a gene or any non-coding sequence (formerly mislabelled as junk DNA). So it may be either a gene, a contig of DNA, or an entire chromosome or genome. In this sequence, we always consider double-stranded DNA as we explore the following three codon reading frames and following the two possible directions of strand reading (3’ ==> 5’ or 5’ ==> 3’). The base unit will always be the triplet codon consisting of three bases.
As shown in above sample, we calculate the Pi-mass related to double stranded triplets DNA bases, double stranded triplets RNA bases, and double-stranded pseudo amino acids. In fact, for each DNA single triplet codon, we deduce the complementary Crick Watson law bases pairing. We do the same work for RNA pseudo triplet codon pairs, then, similarly for amino acids trans lation of these DNA codon couples using the Universal Genetic Code table. Then we obtain 3 samples of pairs codes: DNA, RNA and amino acids and this, systematically even when this DNA region is gene-coding or junk-DNA.
A Simple Example: the Starting Region of Prion Gene:
DNA Image Coding:
ATG CTG GTT CTC TTT...
-1 -1 -1 0 0...
Complement
TAC GAC CAA GAG AAA...
0 -1 0 -2 0...
RNA Image Coding
AUG CUG CUU CUC UUU...
-2 -2 -3 -1 -3...
Complement
UAC GAC GAA GAG AAA...
-1 -1 0 -2 0...
Proteomics Image Coding
MET LEU VAL LEU PHE
4 3 3 4 3...
Complement:
TYR ASP GLN GLU LYS
2 -1 1 0 4...
Pi-masses Corresponding to Two Strands are then Added for Each Triplet
Double strand DNA image coding: -1 -2 -1 -2 0...
Double strand RNA image coding: -3 -3 -3 -3 -3...
Double strand Proteomics image coding: 6 2 4 4 7...
This produces three digital vectors relating to each of the 3 DNA, RNA, and proteomics coded images.
At this point we already reach an absolutely remarkable result, as symbolized in Figure 1.
We will focus now – exclusively - on the DNA code (genomics) and amino acids code (proteomics).
b) The Globalization and Integration Step
To these two numeric vectors we apply a simple globalization or integration linear operator. It will “spread” the code for each position triplet across a short, medium or long distance, producing an impact or “resonance” for each position and also on the most distant positions, reciprocally by feedback. This gives a new digital image where we retain not the values but the rankings by sorting them.
We run this process for each codon triplet position, for each of the three codon reading frames and for the two sequence reading directions (3’ ==> 5’ and 5’ ==> 3’).
For example, to summarize this method: on starting area of the GENOMICS (DNA) code of Prion above, the “radiation” of triplet codon number 1 would propagate well:
-1 -2 -1 -2 0... ==>
-1 -3 -4 -6 -6...then, we cumulate these values: -20
So we made a gradual accumulation of values.
The same operation from the codon number 2 produces:
-1 -2 -1 -2 0... ==>
-2 -3 -5 -5...then, we cumulate these values: -15 etc.
Similarly, the same process on starting area of the PROTEOMICS code of Prion above, the “radiation” of triplet codon number 1 would propagate well:
6 2 4 4 7... ==>
6 8 12 16 23...then, we cumulates these values: 65
So we made a gradual accumulation of values.
The same operation from the codon number 2 produces:
6 2 4 4 7... ==>
2 6 10 17...then, we cumulate these values: 35 etc.
Finally, after computing by this method these “global signatures” for each codon position at Genomics and Proteomics levels, we sort each genomic and proteomic vector to obtain the codon positions ranking: example: as illustrated bellow, the Genomics ranking patterned signature is 2 1 4 3 5 for this Prion starting 5 codons mini subset sequence of 5 codons positions (arbitrary values). Then, to summarize the Master Code computing method on these 5 codon positions starting Prion Protein Sequence
Genomics Signature
Codon 1
Codon / Basic codes / Potentials (with circular closure) / circular complements:
!
-1 -2 -1 -2 0
-1 -3 -4 -6 -6
0
Cumulates: -20
Codon 2
Codon / Basic codes / Potentials (with circular closure) / circular complements:
!
-1 -2 -1 -2 0
-2 -3 -5 -5
-6
Cumulates: -21
Codon 3
Codon / Basic codes / Potentials (with circular closure) / circular complements:
!
-1 -2 -1 -2 0
-1 -3 -3
-4 -6
Cumulates: -17
Codon 4
Codon / Basic codes / Potentials (with circular closure) / circular complements:
!
-1 -2 -1 -2 0
-2 -2
-3 -5 -6
Cumulates: -18
Codon 5
Codon / Basic codes / Potentials (with circular closure) / circular complements:
!
-1 -2 -1 -2 0
0
-1 -3 -4 -6
Cumulates: -14
Final rankings:
Codon positions: 1 2 3 4 5
Potentials: -20 -21 -17 -18 -14
Rankings: 2 1 4 3 5
Then we run similar computing for Proteomics...
Codon 1
Codon / Basic codes / Potentials (with circular closure) / circular complements:
!
6 2 4 4 7
6 8 12 16 23
0
Cumulates: 65
Codon 2
Codon / Basic codes / Potentials (with circular closure) / circular complements:
!
6 2 4 4 7
2 6 10 17
23
Cumulates: 58
Codon 3
Codon / Basic codes / Potentials (with circular closure) / circular complements:
!
6 2 4 4 7
4 8 15
21 23
Cumulates: 71
Codon 4
Codon / Basic codes / Potentials (with circular closure) / circular complements:
!
6 2 4 4 7
4 11
17 19 23
Cumulates: 74
Codon 5
Codon / Basic codes / Potentials (with circular closure) / circular complements:
!
6 2 4 4 7
7
13 15 19 23
Cumulates: 77
Final rankings:
Codon positions: 1 2 3 4 5
Potentials: 65 58 71 74 77
Rankings: 2 1 3 4 5
Then finally:
Codon position: 1 2 3 4 5
Genomics vector: 2 1 4 3 5
Proteomics vector: 2 1 3 4 5
To complete, the same work must be also operate on each codon reading frame.
Meanwhile, a more synthetic means to compute these “long range potentials” for each codon position is the following formula:
Cumulate potential of codon location “i”
Then finally,
Example for Genomics image of codon “i”
The initial computing method described above provides:
-1 -2 -1 -2 0... ==>
-1 -3 -4 -6 -6...then, we cumulate these values: -20
becomes, using this new generic formula:
(-1)x5 + (-2)x4 + (-1)x3 +(-2)x2 +(0)x1 = (-5) + (-8) + (-3) + (-4) + 0 = -20
c) The Great Unification between Genomics and Proteomics Master Code images:
When applying the process described above in any sequence – gene coding, DNA contig, junk-DNA, whole chromosome or genome - a second surprise appears just as stunning as that of RNA neutral element. We find that for one of the three reading frames of the codons given, the Genomics patterned signature and the Proteomics patterned signature are highly correlated.
Contrary to the three genomics signatures which are correlated in all cases, the proteomics signatures are correlated with genomics signatures only for one codon reading frame, and generally in dissonance for the two remaining codon reading frames. Also, there are perfect local areas matching’s focusing on functional sites of proteins, hot-spots, chromosomes breaking points, etc.
In this global correlation, specific codon positions were a perfect match. This is remarkable when regions correspond to biologically functional areas: hot-spots, the active sites of proteins, breakpoints and chromosome fragility regions (i.e., Fragile X genetic disease), etc.
Exmples
-III-Binary code
Function
We compute first-order differential texture and roughness analysis (Leibniz) on Master Code Genomics and Proteomics images.
Inputs
Genomics and Proteomics images data from step II- (2 numerical integer numbers vectors).
Outputs
2 binary vectors (0/1) related Genomics and Proteomics textures analysis.
Summary
If this work is carried out for Genomic patterned pictures, we see that if this trend seems self-organized around one attractor for DNA double strand (Genomics), it shows two levels, two “attractors” for the second (Proteomics). A curious fact then emerges: although two genomics and proteomics curves are still highly correlated in their respective forms and shapes, we discover that their textures are radically different.
Thus the population of Genomics curves will be relatively dispersed around one single withdrawing attractor in a kind of Gaussian dispersion, while the population of Proteomic curves will be distributed around two attractors, bringing out a kind of binary frequency modulation.
We are witnessing the emergence, the “birth” of a Binary Code as demonstrated by Figure 8 , Figure 9, Figure 10, and particularly 3D!
Let us not forget that the initial information was the atomic mass of each bio-atom, which is... a real (decimal) number! Then it is transformed into a code which is an integer number... and it now emerges Binary Code, then 0/1 bits which are binary numbers!
Preliminary analysis shows that the average levels of these two attractors are around 0.61 (61%) and 0.30 (30%) then appear to be in a ratio of two. We will return to these two values bellow.
Here is a small example of a sequence of 312 bases where genomics (red) and proteomics (blue) signature (amino acids) are studied. Note the beauty of these mathematical structures which always increase and that some compare to artistic works by M.C. Escher or J.S. Bach.
Process
Towards discrete Waveforms and logic Biobits overlapping whole chromosomes and genomes
Here we analyze the texture, that is to say, the “roughness” of genomic and proteomic signatures provided by the Master Code. For this, we need only to analyze the slopes or mathematical differentiations from these patterned curves: slopes and gradients - in the sense of LEIBNIZ? - of order 1.
The curves of the Master Code are discontinuous (each point represents a position of triplet codon).
If we note M (i) the Master Code function as defined in -II-, then we agree that:
slope = 1 = ”growing” i.e., “increase” if M (i + 1) > M (i) and slope = 0 = “decreasing” i.e., “decrease” if M (i + 1) < M (i).
Biobits
The emergence of a “binary language” from the Proteomics Master Code of any DNA sequence.
A detailed analysis of the texture of Genomics and Proteomics curves reveals a strange phenomenon: as shown in Figure 8 , Figure 9, Figure 10, a curious roughness or “sawtooth” usually characterizes these images. This somehow amounts to a search for the “derivative of order 1”, that is to say the slope between two successive points. It becomes apparent that these slopes are mostly in the same direction: always growing or always decreasing.
Examples
-IV-Undulatory code
Function
Building discrete waveforms from Genomic Master Code (-II-).
Inputs
Genomics and Proteomics images data from step II- (2 numerical integer numbers vectors).
Outputs
descrete waveforms related Genomics textures analysis.
Summary
Discrete Waveforms: The emergence of “a modulated waveform code” from the Genomics Master Code of any DNA sequence: the generalization of previous gradient differentiations from second, third or nth gradient differentiation order now highlight “bits”… But waveforms, more precisely discrete waveforms of which we will measure periods: period of short-wave or 2 or 3 or even medium-wave wavelengths (greater than 10 times).
Process
Thus, we calculate exhaustively all successive gradients or slopes: S(i, i + 1), and S(i, i + 2), S(i, i + 3), ... S(i, i + n). From all these successive gradients periodicities emerge.
Figure 11 shows shortwave period = 4 codons, then 12 bases pairs in one million base pairs within human chromosome 3. Figure 12 shows long wave period = 12 codons, then 36 base pairs in the first 300000 base pairs within one of the largest human genes, the gene for the genetic disease Duchenne DMD. Figure 12
Examples
-V-Cytogenetic Code


Function
He combination of Proteomics binary code and Genomics undulatory code highlights the light / gray / dark bands characteristic of the kariotypes of each chromosome of the human genome.
Inputs
Proteomics Binary code (-III-) and Genomics undulatory code (-IV-).
Outputs
Kariotypes alternated bands of human chromosomes.
Summary
One of the experimental concrete chromosome representations is a universally known Karyotype image Figure 14). However, the synthesis of two earlier codes (binary code and waveforms) allows prediction throughout the whole human genome of alternating black/gray/white bands of karyotypes as demonstrated by Figure 13. This is the clearest proof of the functional reality of our Master Code discovery. We must recall here that karyotypes are obtained by interferometry, physical process of wave nature.
Figure 13. Example of chromosome 8 bands evidence from Proteomics Binary code and Genomics undulatory code (30 first millions bases in human chromosome 8).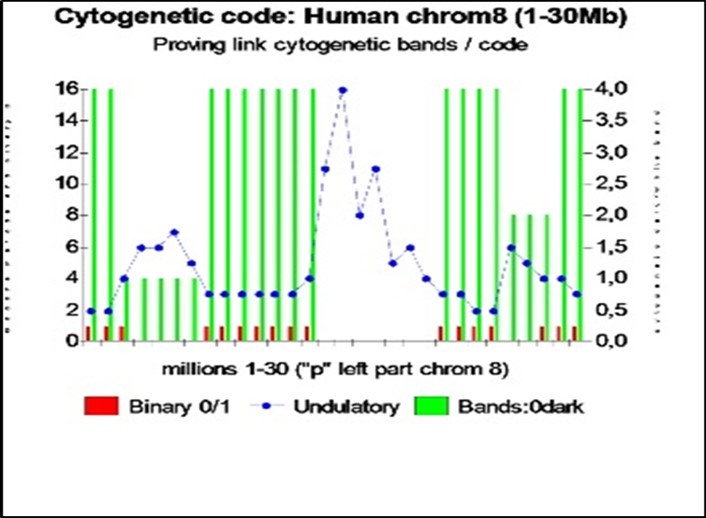
Figure 14. Human chromosome 8 bands of the 30 first millions bases.. 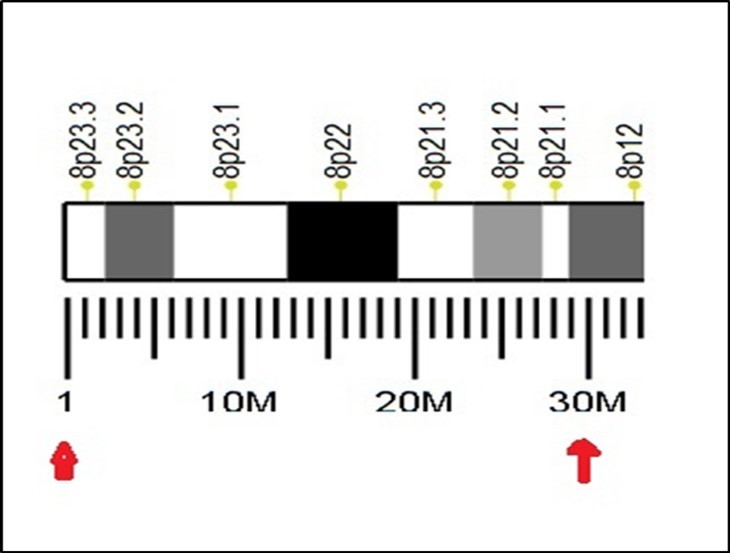
Process
Figure 13 and Figure 14 show an illustration of the “calibration” in a portion of human chromosome 8 (30 first millions of bases).
In Figure 13, the green bars represent the actual referenced colors of the karyotype bands for this region of human chromosome 8: 0 = black, 1 = dark gray, 2 =medium gray, light gray = 3, and 4 = white. Blue curve, modulation of waves is predicted by the textures of the “Master code”: there is a fairly good correlation with the actual referenced colors of karyotypes, particularly, low wave period (2 or 3) for karyotypes clear (white), and waves heavy periods (in this case up to 16) for dark karyotypes (black).
Finally, Biobits, shown here in red, have a status = 1 = “increase” for karyotypes clear (white) and a status = 0 = “decrease” for dark karyotypes (black).
Examples
Such predictive analysis was performed for 24 chromosomes representing the whole human genome. The results show a perfect correlation between the predictions from the textures of the Master Code and grayscale karyotypes as they have been highlighted by the global community of geneticists. Both Figure6a and Figure 6b below show a graphical summary of texture modulations (Genomics) and Biobits (proteomics) throughout the whole human genome. Notes
In each graph, the base unit analyzed in X (horizontally) is the million base pairs: 3266 units representing 3.266 billion bases. Of these, 3075 million bases are significant, while the remaining 191 million relate to GAPs (indeterminate "N" bases), especially the centromeric regions of chromosomes.
The vertical lines delimit the boundaries between chromosomes as well as their centromere regions.
The 2 variations represented correspond respectively to the DNA textures (Genomics) and the amino acid textures (Proteomics). They are calculated independently for each of the millions of bases analyzed, ie "one point" per million bases on the genomics curve and "one point" per million bases on the proteomics curve.
Although the two Genomics and Proteomics variation curves are very strongly correlated (96.63% on average throughout the genome), their respective "textures" are radically different!
In fact, the GENOMICS texture is "ANALOGIC modulated" around an average value close to 60% (graduation 6000) ... which would seem to be phi = 0.618.
On the other hand, the PROTEOMICS texture (although calculated in exactly the same way and on highly correlated curves), is "modulated according to a BINARY LOGIC", oscillating permanently between 2 attractors whose respective values are: Floor = FLOOR = 30% on average, or Ceiling = CEILING = 60% on average. The ratio between these 2 attractors is therefore very close to the number "TWO". The "clouds" of points perfectly illustrate the reality of these two 0/1 bit attractors or "FLOOR / CEILING".
Figure 15. The first 8 Chromosomes (1 to 8) of the whole HUMAN GENOME.
Figure 16. The 16 other remaining Chromosomes (1 to 8) of the whole HUMAN GENOME.
-VI- Standing Waves Meta-Code
Function
The Genomics master code (-II-) is generalized to meta-codons that no longer have 3 nucleotides as a codon, but 4, 5, ... 377 nucleotides. Then we analyze the textures by the undulatory code (-IV-). It then appears dissonances and resonances that will reveal periods of discrete waves, resonances, and standing waves. The Genomics Binary code analysis (-III-) confirms these periods using a complementary independant method.
Inputs
Double strand DNA sequence Pi-mass grouped by meta-codons (each Pi-mass is = -1 times number of « G » bases in meta-codon double strand or also = -1 times number of « C+G » bases in single strand meta-codon.
Outputs
Peiod and resonance standing wave computed by two complementary methods.
Summary
We introduce here a method of global analysis of the roughness or fractal texture of the DNA sequences at the chromosome scale. To do this, we generalize the method of numerical analysis of the "Master Code" (-II-). Thus, we restructure the sequence into different generic sequences based on "meta codons", no longer triplets of 3 nucleotides, but values ranging from 17 to 377 nucleotides, ie 360 simulations. This method of analysis will then reveal, in most cases, discrete waves or interferences, most often dissonances (based on Genomics Undulatory waves described here in -IV-). However, sometimes there will emerge kinds of resonances where all scales of analysis appear to be in symbiosis.
Process
The discrete interferences fields resulting from the analysis of an entire chromosome are therefore a three- dimensional space: Dim y (vertical) restructuring in meta codons of lengths 17 to 377 nucleotides Dim x (horizontal) Leibnitz differentiations such that prmary 1/2 secondary 1/3... 1/4 ... 1 / n Dim z cumulated populations from the "Master code" operators. The + 1/ -1 derivatives will be of type increase, ie +1 if derivative increasing and will be of type decrease, ie -1 if derived decreasing. In this context we will explore these 3D spaces in 2 forms:
Horizontally (IV- Undulatory code), meta codons dimension: curves for a given meta codon dimension, see in the example "resonances" below (see Figure 17 and Figure 18).
Vertically (-III- Genomics binary code), spectral differentiation: discrete series d2-d1 is +1 if increase and -1 if decrease (see Figure 19). We represent in top the +1 and in low the -1, (see Figure 19). Table 5
Dim x d1 d2…/... d100
Dim y …/... 377
Horizontal Scan : exp. meta codons of 22 bases : 22 761233 774174 779102 783714 786854 …/...
(see Figure 17)
Vertical Scan : example derivations of first order: 1 if d2>d1 and -1 if d2<d1 then : -1 1 -1 1 -1 1 1 -1 1 -1 1 1 …/...
These two independent methods lead in all the cases analyzed to the same period value: here, for example, the period "horizontal scan" is a resonance of 22bp (Figure 18) and the period "vertical scan" is a period of repeatability of 22bp also (Figure 19).
Figure 17. Zoom on vertical scan method revealing PERIOD = 22 from HG38 reference chromosome21.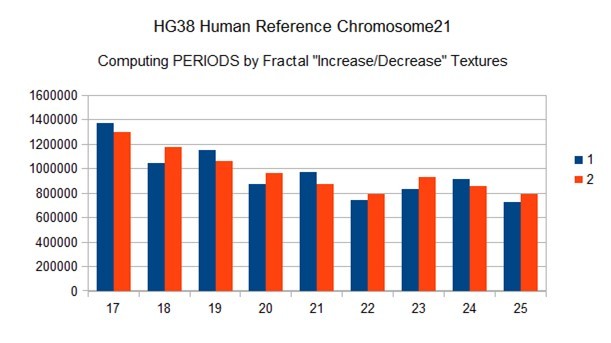
Figure 18. Evidence of a resonance of 22bp period in the whole HG38 human reference chromosome21 (horizntal undulatory code -IV-).
Figure 19. Confirmation of a 22bp period in the whole HG38 human reference chromosome21 -vertical genomic binary code -III-)
A third complementary method is presented here: knowing the period determined and confirmed by the two previous methods, we segment the complete sequence of the chromosome by consecutive segments according to this period, for example here for the chromosome21, we will "cut" the entire sequence of the chromosome in successive sections of 22 bases, the length of the period discovered. Then we record for each segment the C + G populations on the one hand and T + A on the other hand. We then represent the cumulative distribution curve of these different CG and TA populations throughout the chromosome sequence. Table 5 segmented by 22 bases periods.
Results and Discussion
First Experiment: “Natural Hierarchical Introspective Logics”
We revisit here "HIERARCHICAL INTROSPECTIVE LOGICS" The theories of John F. NASH 3 by highlighting the self-emergence of a bistable logic. structuring the entirety of the human genome (Logic code -III- in §Methods).
“There can exist no procedure for finding the set of all regularities of an entity. But classes of regularities can be identified. Finding regularities typically refers to taking the available data about the entity, processing it in some manner into, say, a bit string, and then dividing that string into parts in a particular way and looking for mutual AIC (Algorithmic Information Content) among the parts. If a string is divided into two parts, for example, the mutual AIC can be taken to be the sum of the AIC's of the parts minus the AIC of the whole. An amount of mutual algorithmic information content above a certain threshold can be considered diagnostic of a regularity. Given the identified regularities, the corresponding effective complexity is the AIC of a description of those regularities”.
In one hand, in his unformal paper entitled “HIERARCHICAL INTROSPECTIVE LOGICS” 3, the Economy Nobel prize the mathematician Professor John F. Nash Jr. explores new approaches of Turing/Godel undecidability problems adding particularly the “EMBEDDABILITY” dimension.
In other hand, we report here mathematical CODES structuring all genomes and particularly the whole sequenced Draft Human Genome 9 released in 2001 (three billions base-pairs about distributed in the 24 Human chromosomes). This discovery describes particularly the evidence of a self-emerging embedded BINARY CODE structuring the whole human genome.
Then, analysing the texture (mathematical increase/decrease 1st degree derivates) of the Proteomics (code -III- in §Methods) associated curve patterned signature, we could associate with each codon position “biospins” as following:
If the local codon position derivate is in increase state è then Biospin=1,
If the local codon derivate derivate is in decrease state è then Biospin=0.
Now, for any analysed sequence, we could compute the related balancing increase/decriease percentage related to the whole analysed sequence. These percentage values are real numbers in the range 0-100. Normally, the distribution of biospins percentages must be random, like a Gauss-like distribution.
In fact, we obtain a very strange distribution as a “bath-tub”-like distribution: there appears, in ALL CASES, a binary distribution centered around two ATTRACTORS: one attractor, named “Floor-state attractor” is located around 29%.
The other second attractor, named “Ceiling-state attractor” is located about around 60%.
The following Law is Universal. We Propose the Following Rule Entitled “Genomic BINARY CODE Law”
For any sequence " seq " of genomic DNA, what ever its length, its position, and its nature, one can always associate, by applying the numerical algorithm described in (code -III- in §Methods), a Binary Code status, called « BioBit » such as:
BioBit (seq) = 0 = « Floor » state = « FALSE » if %(seq) neighbouring attractor 29%.
BioBit (seq) = 1 = « Ceiling » state = « TRUE » if %(seq) neighbouring attractor 60%.
We validated and checked this universal law on the totality of the genomes known to date, and, more particularly, on the whole human genome which we studied independantly on three embedded scales: contiguous segments of 10000bases, 100000bases and 1million of bases from Build34 2003 Human genome release 10, see Figure 15 and Figure 16 in §Methods.
We demonstrate now this Nash's sugested law “Natural Hierarchical Introspective Logics” on a randomly selected region within the Draft Human Genome sequence 9. This genomic studied region is located between 130000000 and 131024000 positions within the human chromosome5.
Some regions are undefined (“N” undefined bases or “GAPS”).
We Run 11 Independant Embedded Analyses:
-1024 contiguous DNA segments of 1000bases.
- 512 contiguous DNA segments of 2000bases.
- 256 contiguous DNA segments of 4000bases.
- 128 contiguous DNA segments of 8000bases.
- 64 contiguous DNA segments of 16000bases.
- 32 contiguous DNA segments of 32000bases.
- 16 contiguous DNA segments of 64000bases.
- 8 contiguous DNA segments of 128000bases.
- 4 contiguous DNA segments of 256000bases.
- 2 contiguous DNA segments of 512000bases.
- 1 unique DNA segment of 1024000bases.
In the Following Table, we Resume, for the 11 Independant Analyses:
1. The numbers of elementary BioBits decisions: exp in line 1: 468 “Floor states” and 409 “Ceiling states”, the total correspond to 877 segments, the remaining are GAP segments.
2. The average values of Floor and Ceiling percentages: exp in line 1: 31% for “Floor attractor” and 59% for “Ceiling attractor”.
3. The LOCAL LEVEL VOTE DECISION: exp in line1, the Floor state (468) is majority then the local level decision is “Floor”=FALSE. Table 6
In the Following Couples of Graphics we Demonstrate the SCALE INVARIANCE and the EMBEDDED LOGICAL VOTE Process
è Level 1: 1024 times 1000 bases... Concensus Decision “VOTE” = Floor = “False”.Figure 20, Figure 21, Figure 22, Figure 23, Figure 24, Figure 25, Figure 26, Figure 27, Figure 28, Figure 29, Figure 30, Figure 31
Level 2: 512 times 2000 bases... Concensus Decision “VOTE” = Floor = “False”
è Level 3: 256 times 4000 bases... Concensus Decision “VOTE” = Floor = “False”
è Level 4: 128 times 8000 bases... Concensus Decision “VOTE” = Floor = “False”
è Level 5: 64 times 16000 bases... Concensus Decision “VOTE” = Floor = “False”
è Level 6: 32 times 32000 bases... Concensus Decision “VOTE” = Floor = “False”
è Level 7: 16 times 64000 bases... Concensus Decision “VOTE” = “Undefined”
è Level 8: 8 times 128000 bases... Concensus Decision “VOTE” = Floor = “False”
è Level 9: 4 times 256000 bases... Concensus Decision “VOTE” = Floor = “False”
è Level 10: 2 times 512000 bases... Concensus Decision “VOTE” = Floor = “False”
Figure 30 - Level 10: 2 times 512000 bases... Concensus Decision “VOTE” = Floor = “False”
Figure 20. Gauss like CG / TA distribution within the whole human HG38 chromosome21
Figure 21. Level 1: 1024 times 1000 bases... Concensus Decision “VOTE” = Floor = “False”
Figure 22. Level 2: 512 times 2000 bases... Concensus Decision “VOTE” = Floor = “False”
Figure 23. Level 3: 256 times 4000 bases... Concensus Decision “VOTE” = Floor = “False”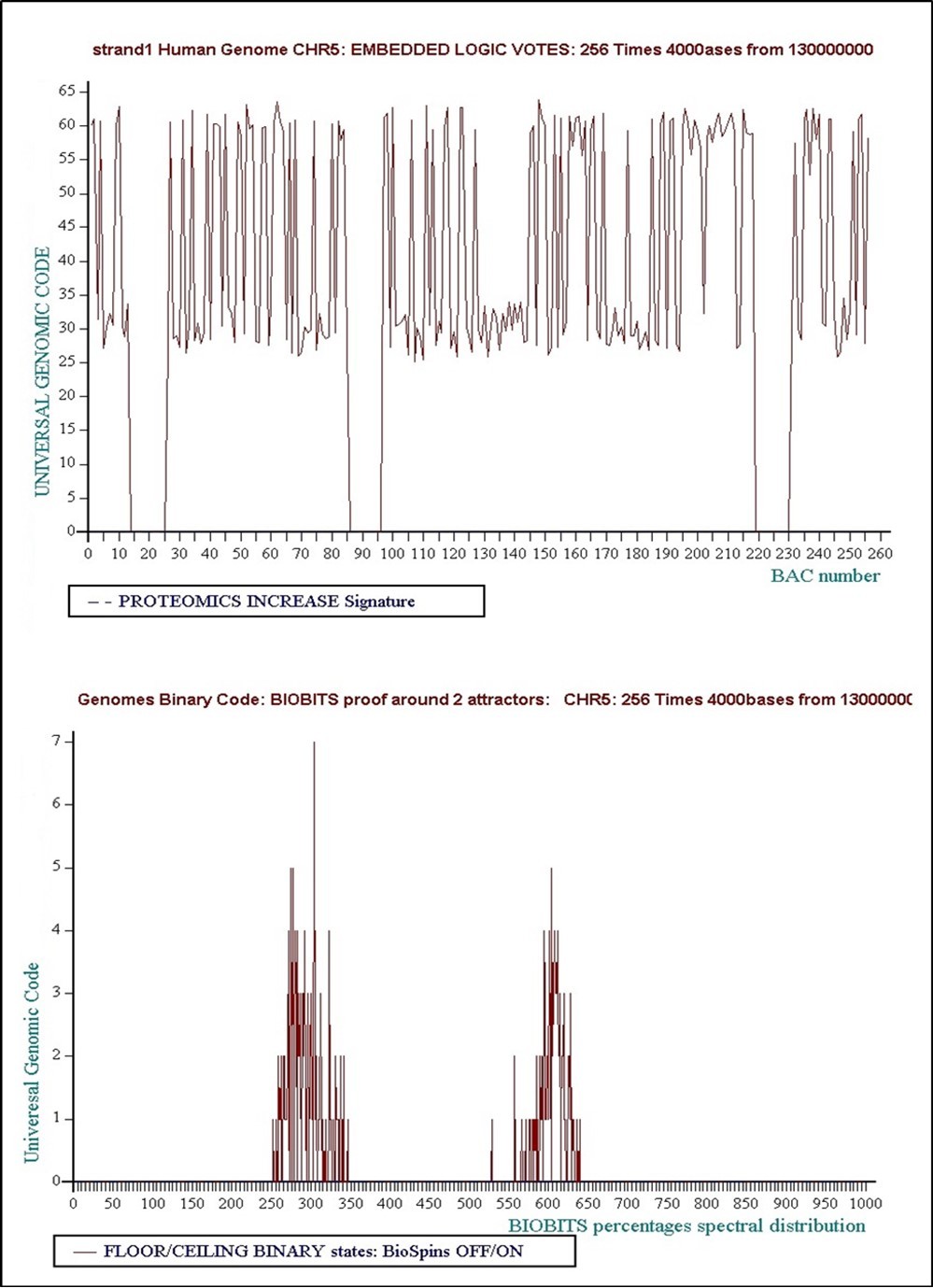
Figure 24. Level 4: 128 times 8000 bases... Concensus Decision “VOTE” = Floor = “False”
Figure 25. Level 5: 64 times 16000 bases... Concensus Decision “VOTE” = Floor = “False”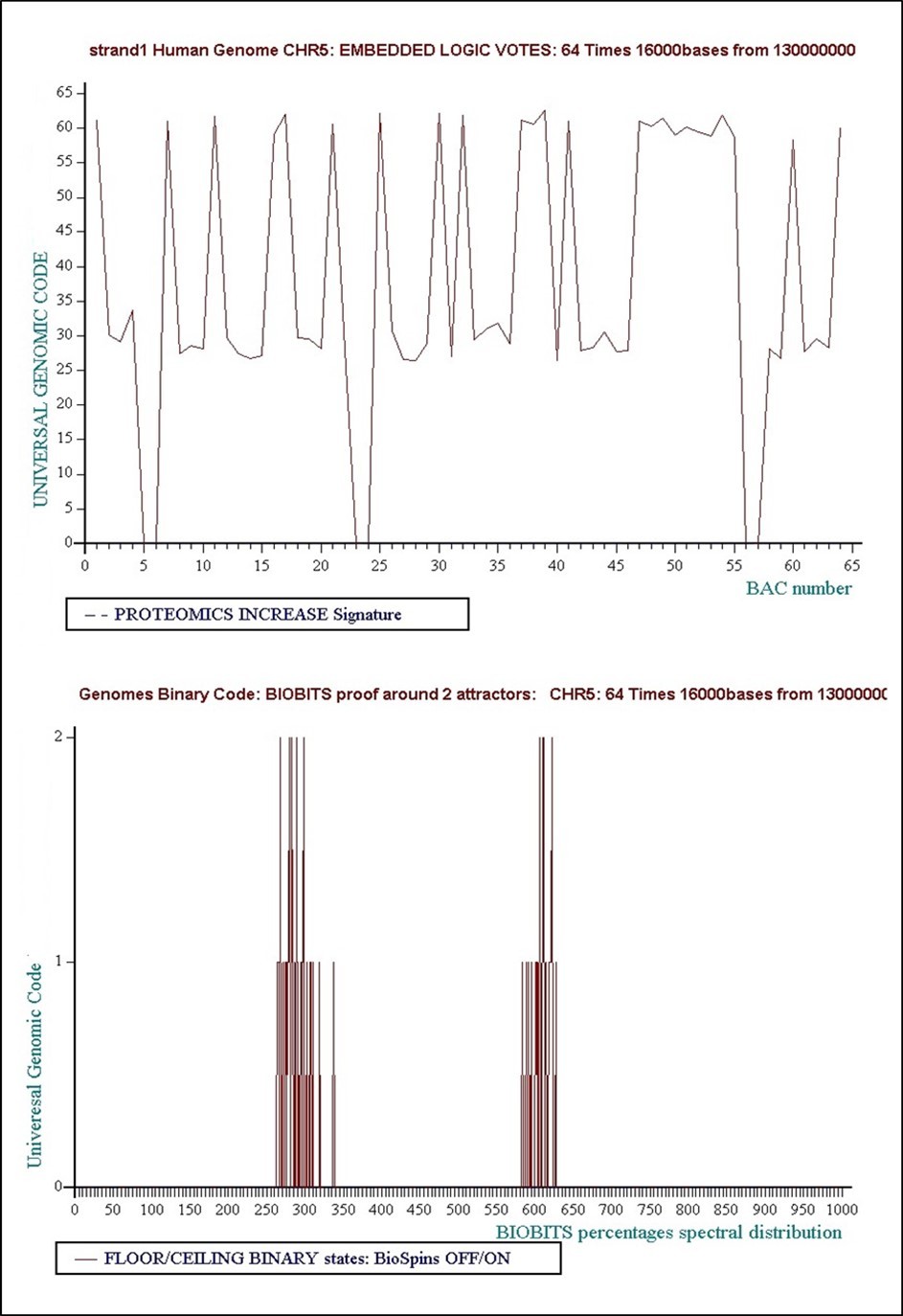
Figure 26. Level 6: 32 times 32000 bases... Concensus Decision “VOTE” = Floor = “False”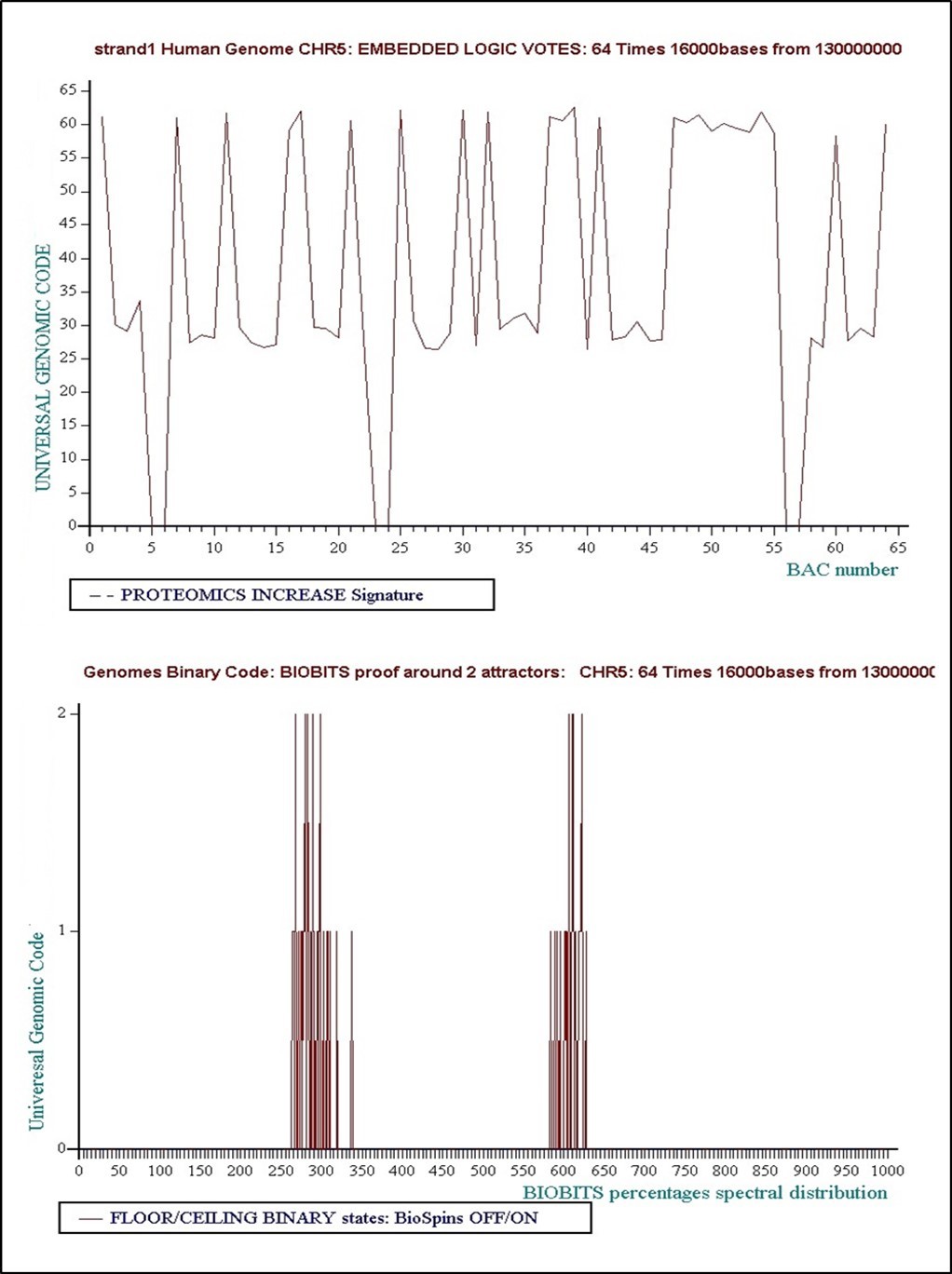
Figure 27. Level 7: 16 times 64000 bases... Concensus Decision “VOTE” = “Undefined”
Figure 28. Level 8: 8 times 128000 bases... Concensus Decision “VOTE” = Floor = “False”
Figure 29. Level 9: 4 times 256000 bases... Concensus Decision “VOTE” = Floor = “False”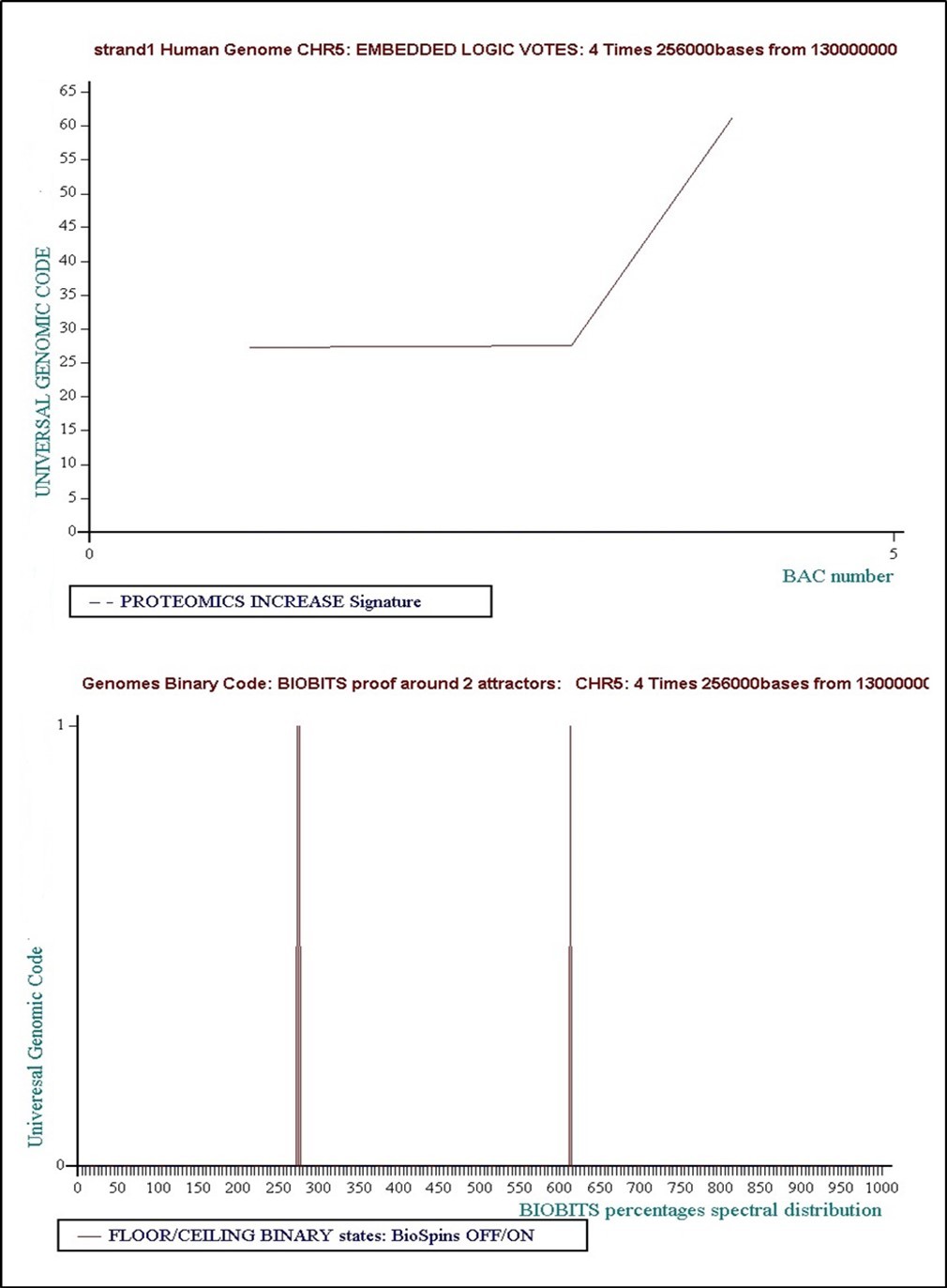
Figure 30. Level 10: 2 times 512000 bases... Concensus Decision “VOTE” = Floor = “False”
Figure 31. 10Millions bases genomic region around the 1024000 bases analysed region is globally also in “floor” binary state.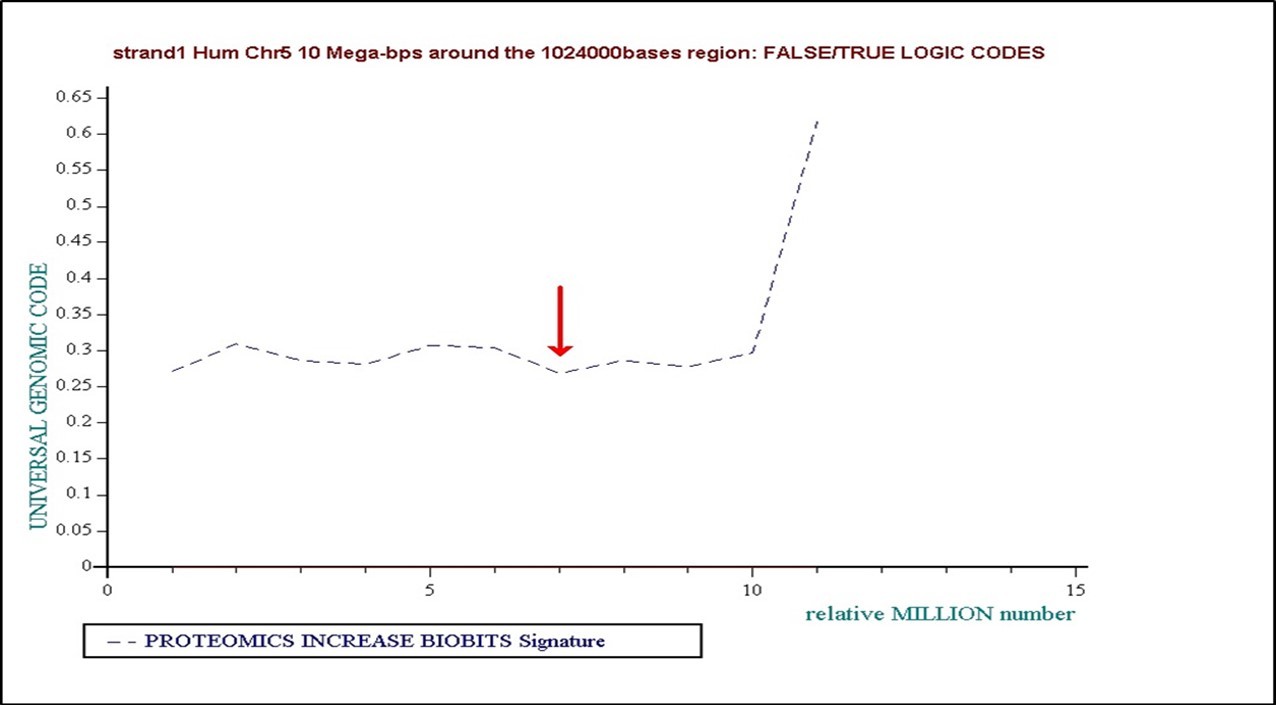
In the following graphic, we summarize the genomic area around the 1024000bases DNA sequence analysed. This 10 millions lenght sequence was analysed spliting it in 10 “ONE MILLION regions”. The red arrow localizes the studied 1024000 studied region. Then, the area is globally at “Floor” state, with a “Ceiling” state transition at the end (see on the right).
As Professor NASH in " Hierarchical Introspective Logics “Proposes it:
On the one hand, the concept of incompleteness will be able to evolve with the levels of human knowledge. In addition, the discovery of new natural laws will be able to make evolve the approach of this problem “.../... But the history of human progress in science and mathematics reveals that observation of the phenomena of Nature has always played a large rôle.../...” (by J. F. NASH in the above paper).
In other hand, the concept of LOGIC suggested here is radically new because it acts of a self-emerging logic, output of a self-organized multi-levels embedded process of which the roots (" ground level ") are at the basic level of the “average atomic masses of the 6 DNA CONHSP bio-atoms”, therefore in a world of real numbers (atomic mass code -I-).
Lastly, the concept of hierarchy is not discrete but completely continue in an infinity of embedded levels. Thus, the choice of 11 levels in the example suggested is arbitrary, one could also have chosen thousands of others embedded levels... We show thus that the human genome (as all the other genomes) is the source of an omnipresent logical binary language which appears to be invariant on all the scales. This code is not explicit and formal but self-emergent as the " output " of a complex genetic system. This code is embedded in a self-referred infinity of VOTE-like level. Perhaps it could give new ways and tracks to understand the “Natural Hierarchical Intro spective Logics” decision making process.
Second Experiment: “Proof of the Optimality of the Life Isotopes Atomic Masses Proportions Fine Tuned Balance”
Thus, the totality of the average atomic masses of all basic materials of genetics, bioatoms, UTCAG bases, amino acids, codons, DNA RNA and proteins are unified as soon as they are mathematically projected in the space of PIs. masses (Atomic mass code -I-).
But why their average atomic masses rather than the atomic masses of individual isotopes?
Does this property extend to the isotopes constituting their bioatoms?
The Meta-structure of Isotopes
A new question naturally arises:
"Would the projection of the PI-masses be" reserved "and exclusive to the atoms of the living? "
"What would happen if we applied this same projection to all the other atoms of nature? "
We therefore apply the law of projection of PI-masses to all known stable isotopes. This list is then extended after the isotope Bi209 which follows the radioactive isotopes of the following elements. We then discover two remarkable phenomena:
on the one hand, a periodicity = 3.
On the other hand, a symmetry which would be located on both sides of the Lanthanides region (more precisely towards the element CE140.
Finally, it should be noted that these meta-structures are destroyed by a deliberate random disturbance of the atomic masses of all these elements of the order of 1/1000).
Symmetry of the PI-masses of the 306 successive isotopes of the Mendeleev Elements table.
Figure 32 above illustrates very well the symmetry between the 306 isotopes revealed by the projection PI-masses. This symmetry is located on both sides of the Lanthanide element zone; the sudden break between regular elements and radioactive elements (inflection of the graph, see blue arrow) located on the right of the graph corresponds to the transition between regular elements and elements (towards element Bi209).
Proof of the Optimality of the Average Atomic Masses
In figure 33 below, we study the PI-masses in the case of bioatoms or most basic molecules. Those who are the origin of life and who maintain life on earth. But also those that can be encountered in outer space: clouds of interstellar dust, atmosphere of TITAN etc ...
We will analyze here among others carbon C, oxygen O, primitive molecules NH, N2, H2, H2O (water), CO, CO2 (carbon dioxide), CH3OH, and NH3. Let 5 organic atoms and 5 interstellar molecules. In each of these 10 cases, we will analyze the PI-masses of the molecule formed by the lightest isotopes, the average atomic masses, and the heavier isotopes. Thus, NH will be formed
1. In the first test of N14 and H1 (the lightest isotopes of nitrogen and hydrogen).
2. In second test of N and H represented by their average atomic masses.
3. In the third test of N15 and H3 (heavier isotopes of nitrogen and hydrogen). and so on for each of the 10 tests of molecules analyzed.
In Figure 34 and Figure 35, we now study in a similar way a patchwork of several thousand bio-molecules essential for life, from the most primitive bioatoms, to the amino acids, and through the 11 bio-molecules citric metabolic control, all are essential organic compounds for life. All these molecules are derived, on the one hand, from primitive compounds such as can be found in interstellar clouds, and on the other hand, from basic organic compounds extracted from the famous BEILSTEIN 11 ranging from compounds simple to more complex compounds (bases and amino acids).
In this first type of simulation, we submit 12 test sets of "more or less" organic molecules to a set of 3 tests: in the middle, the actual and exact average atomic mass, and, on both sides, on the left. , this same value weakened by a ten thousandth, and on the right, the same value, increased by a ten thousandth. The compared values measure the error observed when adjusting the PI-masses of these various atomic masses by the associated PI-mass integer (multiple of Pi/10). As a result, the higher these values, the poorer the PI-mass optimality. It is therefore concluded that these minute random "noises" applied to the atomic masses destroy the optimality of the PI-mass nPi/10 image value, hence, consequently, the optimality of the atomic mass as well.
On the other hand, the more the organic compounds are to the right, the lower the PI-mass error (average atomic mass), and the more this error becomes hypersensitive to perturbations of its atomic mass. Thus, compounds such as amino acids would be more optimal than interstellar organic compounds, themselves more optimal than non-organic interstellar compounds.
Would the whole path and path of evolution be "trapped" by our discovery of the digital projection of PI-masses?
Finally, a final confirmation of this meta-order revealed by PI-masses is visualized by the analysis of Figure 35 below.
We compare the perturbations of the optimal PI-masses caused, on the one hand, by the substitution of average atomic masses by mono-isotopic atomic masses, and, on the other hand, the substitution of average atomic masses by noisy atomic masses by a tiny random noise. The first disturbance is real and natural whereas the second disturbance is artificial and the virtual order of the simulation.
In all these cases, the reference is the slight thin horizontal line annotated 100%.
We then observe that the more we move to the right of the graph, hence from the inorganic to the organic, and the more the effect of real (isotopic) or artificial (random) perturbations will then increase, become showing strong overtaking above the 100% mark horizontal line.
We conclude the optimality of the average atomic masses vis-à-vis both mono-isotopic atomic masses that atomic masses whose precise value would be very slightly disturbed.
Figure 32. Symmetry of the PI-masses of the 306 successive isotopes of the Mendeleev Elements table.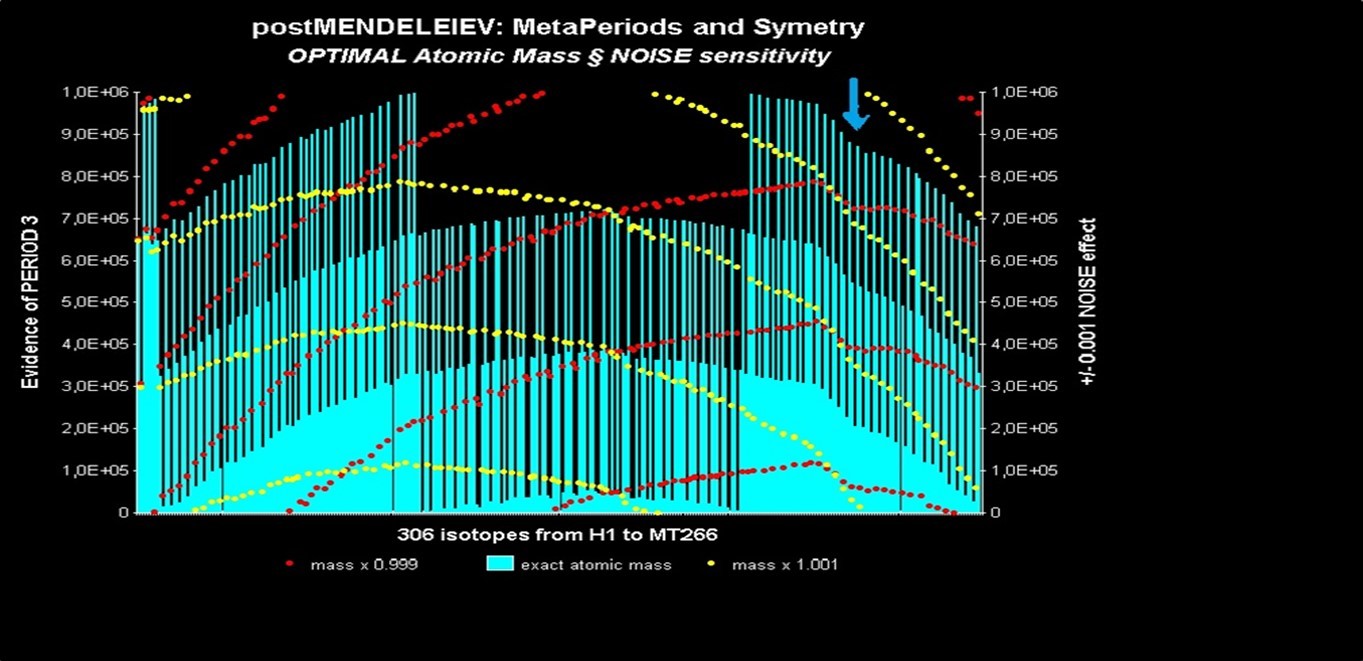
Figure 33. The OPTIMALITY of the average atomic mass is proved here on the PI-masses of ten primordial organic components.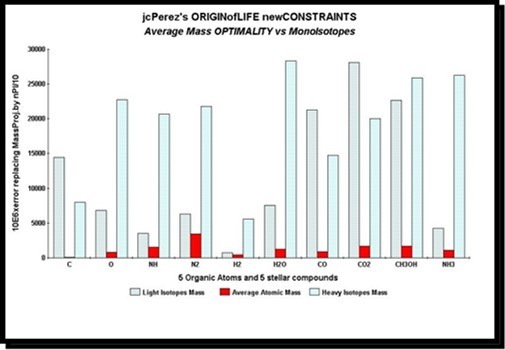
Figure 34. Evidence of noise sensitivity of average atomic masses for 12 sets of organic compounds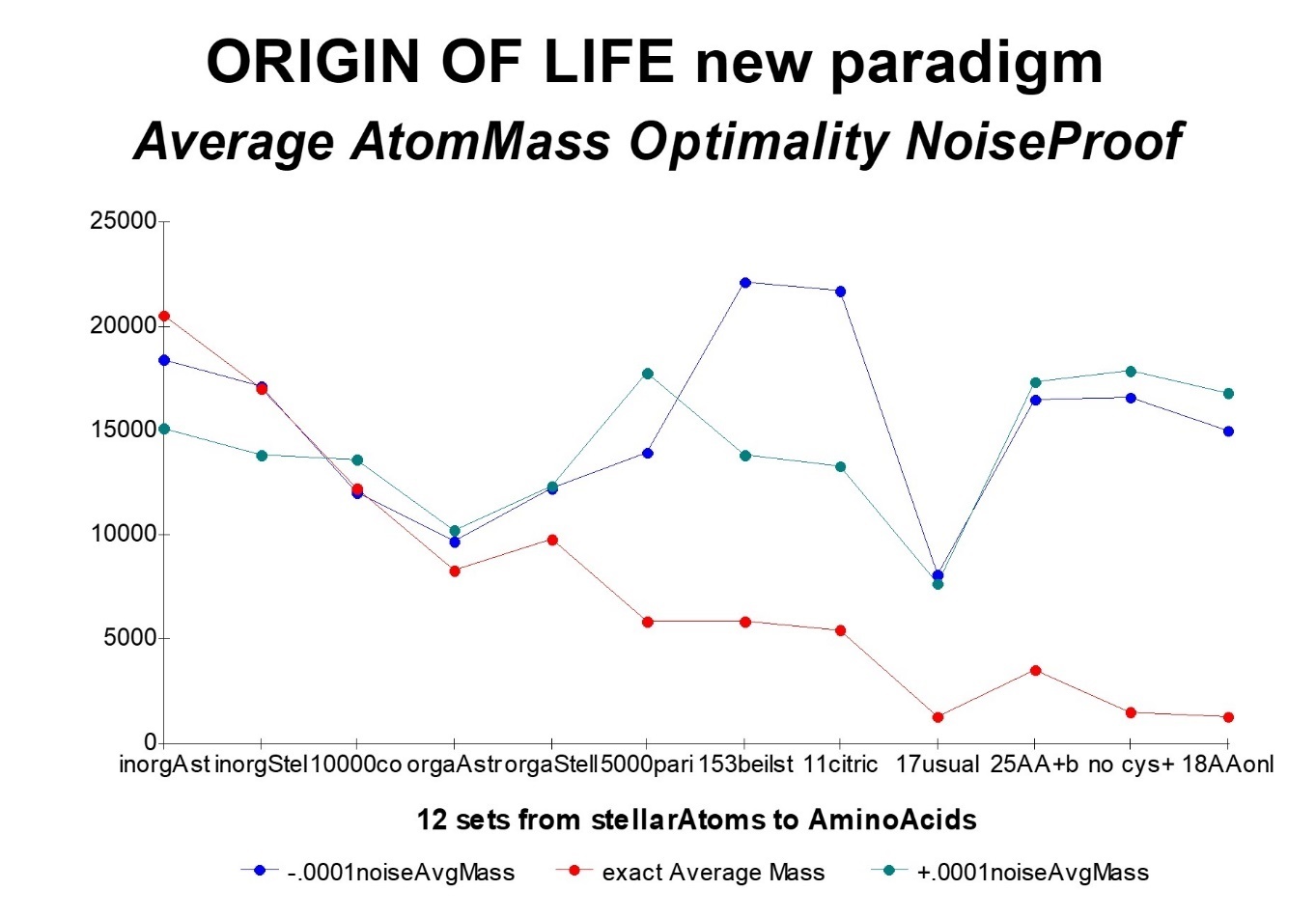
Figure 35. The comparison of the ratios "monoisotopic mass / average mass" between real cases and noisy cases.
Perspectives in Exobiology
Concentrated information and meaning represented by the 3 figure 34, figure 35, figure 36, it will emerge remarkable information: more organic compounds are moving towards more evolution, from small molecules premisses of life, until Amino acids, bricks of all life, and more the selectivity, the optimality of the atomic masses becomes pointed and adjusted, the work of a grandiose "tuning" ... And it is from this PI-mass - perfect - that go animate whole life "codes": PI-masses (atomic mass code -I-)!
Indeed, it is thanks to this hyper-adjustment of PI-masses that will emerge codes and even real languages that will organize and unify ALL genetic information despite its apparent diversity - bioatoms, DNA, RNA, amino acids proteins and genomes - to lead and induce a high organization up to the scale of whole genomes: from the atom to the genome (code -I- to code -VI-).
A mystery however is that these optimal values of the masses atomic ones are those of the average atomic masses and not those of the mono-isotopic atomic masses ... However, let us remember that the average atomic mass is only ... average. That is to say that not a single molecule, not a single nucleotide, not a single amino acid has this keystone value from which come alive the "codes" in whole numbers of life: the PIs -masses! But the reality is that this ideal PI-mass is only a deception, a kind of "Omega Point" Teilhard Chardin that no molecule can ever achieve, at best only some will approach. Thus, we discover an ideal law, which can only exist in a mass universe, probabilistic, a universe of means, a universe of large numbers. In fact, you will never meet a carbon atom whose mass is exactly 12.01114727 (its Carbon average atomic mass). On the contrary, you will encounter a multiplicity of individualized, mono-isotopic atoms: billions of C12 isotopes, other billions - albeit a little less - of C13 isotopes ... In the same way, you will never come across an Oxygen atom of which the mass is exactly 15.99929706 (its Oxygen average atomic mass). On the contrary, you will find a multiplicity of individualized, mono-isotopic atoms: billions of O16 isotopes, other billions - albeit a little less - of O18 isotopes ... or even more rarely, billions of isotopes O17 Here is the famous curve in "V" that we will call by analogy "Sergeant Major curve".
We are therefore Faced with an Enigmatic Situation
On the one hand, we discover a generic law organizing the entirety of living molecules - the PI-masses code (atomic mass code -I-).
On the other hand, this law is optimal only in the case of the average atomic masses.
But the real world of biology is not a world of average atomic masses but a world of atomic masses mono-isotopes.
This would mean that this optimal law is only a kind of "inaccessible star" as Cervantes would have written.
But, nevertheless, the average atomic masses exist on earth since they constitute the weighted average of the billions of isotopes constituting each one of the populations of atoms.
Then would it mean that life implies, by the very nature of its laws, that its molecules and atoms rely on diversity and multiplicity, perhaps even on the equilibrium and collective co-operation between these billions of atoms?
Life through the optimal search for the average PI-masses would imply in every way spaces of life and permanently a kind of permanent dynamic equilibrium Piaget spoke of "mobile equilibrium" - taking its roots and feeding on the multiplicity and diversity of the different isotopes (C12 / C13, N14 / N15, H1 / H2, O16 / O17 / O18, etc.) forming each bioatom. It will be noted that the complexity of advanced molecules such as amino acids already favors this diversity, the same amino acid can be heterogeneous and consists of different isotopes of the same atom, which will already approach some of the PI-mass ideal average mass (for example the amino acid glycine (Gly) which is written NH2 CH2 COOH may for example consist of an atom O16 and an atom O18, likewise for its carbon atoms which may be 2xC12 or 2xC13 or C12C13.
It will also be noted that this multiplicity of isotopes can be topological (3-dimensional space) but also temporal.
Topological diversity and we then think of the GAIA 12 hypothesis of James Lovelock, about which we wrote.
« He is to science what Gandhi was in politics. His central idea, that the planet behaves like a living organism, is as radical, profound, and vast in its consequences as any of Gandhi's ideas. »
According to Lovelock, everything that lives on our planet would contribute more or less to the balance, maintenance and continuity of life on earth. But it turns out that Lovelock as we reach by taking two radically different paths.
Immediate Consequences of this Requirement of Multiplicity of Isotopes are Multiple
The loss of bio-diversity destroys this balance of average atomic masses required by life.
-the discourse on global warming must climate change revisited according to this new angle of the requirement of isotopic equilibrium. We will think of cancers whose hidden causes are more and more due to the industrial pollution of modern life and agro-industrial mass supply.
The economic arguments of certain lobbies according to which the methane released into the atmosphere by the cows of our campaigns would pollute almost as much (1/5) as the methane rejected by the industries of our cities will suddenly fall: indeed we will have to consider seriously the hypothesis that the isotopes constituting CH4 (methane) released by our cows are very different from the isotopes of CH4 released by our polluting industries!
The other level of isotope multiplicity of the same atom is the temporal and dynamic aspect.
This Inspires us with Two Ideas
On the one hand, this could partly explain the natural and permanent need for renewal and re generation of living cells. Nature would thus seek to maintain permanently the isotopic equilibrium of the respective proportions of the billions of isotopes of bioatoms forming the living cells.
On the other hand - it may seem bold as a thesis - the dynamic equilibrium of the average atomic masses can also be reached by temporal alternations of the various isotopes constituting the same atom. It would then be a kind of "quantum dance" with a kind of temporal alternation between isotopes of the same atom ... We would then be very close to the wave or quantum theories of matter ... The real but invisible atom would have as mass the mass atomic average while we perceive in the universe palpable and visible that different "views" with distinct masses, the different isotopes of the same atom.
A bold thesis, of course, but why not?
One thing is certain when exobiologists will look for traces of life on other planets, on TITAN, on MARS or EUROPA ... They will not only have to look for the presence of carbon, nitrogen oxygen or hydrogen, but also the existence of isotopes of these atoms in proportions comparable to those which constitute the average atomic masses on the earth biosphere.
Thus, it is in 2010 that a probe went to analyze with precision the reports isotopic C12 / C13 or N14 / N15 on TITAN ...
Have we discovered proportions comparable to those of our planet?
Only in such a case that our discoveries could allow to predict the isotopic ratios that it should find there ... Then, the projections PI-masses of the code of the atomic masses will be able to measure these traces of extra-terrestrial life.
We will Indeed Affirm that
"The emergence of life in the Universe requires a DIVERSITY and an ISOTOPIC BALANCE of bioatoms C O N H identical to those observed on Earth"
or…
"A necessary but not sufficient condition for the emergence in the universe of life-forms similar to earthly life requires that we find, in these regions of the universe, and in MARS in particular, the different isotopes of organic CONH atoms in proportions identical to those observed on Earth ".
Perspectives in Oncology and Cancer Therapy Basic Reesearch
The biomathematical methods presented above open very wide and promising perspectives in the theoretical study of the mechanisms of emergence, detection and treatment of cancers. Here are 3 remark able examples published in 2017 and 2018.
HGO and Cancer LOH Deletions:
In (Pérez, jan 2018 and March 2018) we demonstrate that HGO is an optimum destroyed by LOH (Loss Of Heterozygosity) involved in all Cancers: “When an LOH deletion affects a chromosome upstream of the HGO point (chromosomes 4 13...) in the chromosomal spectrum, this deletion degrades the genomic optimum of the cancer genome. When an LOH deletion affects a chromosome downstream of the HGO point (chromosomes 19 22...) In the chromosomal spectrum, this deletion improves the genomic optimum of the cancer genome. The exhaustive analysis of the 240 LOHs for the following 6 cases: Chromosome 13 (breast cancer), chromosome 5 (breast cancer), chromosome 10 (glioblastoma cancer), chromosome 1 (colorectal cancer), chromosome 1 (neuroblastoma cancer) and chromosome 16 (prostate cancer) obey this law in 227 cases and do not obey this law for 13 cases (success rate of our law = 94.58%). In (Perez, Jan2018) we detailed this type of analysis on 153 LOH relating to breast and prostate tumors affecting respectively chromosome 13,chromosome 5 (breast) and chromosome 16 (prostate). In this detailed study, the HGO law described here is edified in 143 cases out of 153, or 93.46% of favorable cases." See also in (Perez, March2018) an analog analyse on brain cancers neuroblastoma and glioblastoma (see example below, Table 9).
Table of synthesis of 240 LOH relating to 5 types of cancer and 5 different chromosomes. The shaded areas represent those that comply with the HGO law, ie 227 cases out of 240. In blue the 53 caeses related to Neuroblastoma and Glioblastoma cancers.
The shaded areas represent cases of LOH complying with the generic rule, while the unshaded areas represent cases of LOH that do not comply with this rule. In total, 227 cases respect the rule and only 13 cases of LOH do not respect it, a success rate of this law equal to 94.58%. In particular, on 53 cases of LOH mutations analyzed for the two types of Glioblastoma and neuro blastoma cancers, the HGO optimality law is verified in 50 cases and in failure for only 3 cases, ie a percentage of validity of this universal law equal to 94.24. %. Figure 37
Mt DNA Genomes and Cancers
In (Pérez, 2017) these Fibonacci resonances are used to demonstrate that: “…by analyzing 250 characteristic mutations associated with various pathologies, we establish a very strong formal causal correlation between these numerical Fibonacci meta structures (Figure 6) and these referenced mtDNA human genome mutations involved in cancers or LHON genetic disease (Leber Hereditary Optic Neuropathy)”.
Graphics related 10946 resonances for 36 cases of mutations associated with the LHON disease (Leber's hereditary optic neuropathy). In this figure, the 36Table 5, Table 6, Table 7
LHON mutations on mtDNA genome INCREASES the number of long 10946 Fibonacci“resonances”. Figure 38, Figure 39, Figure 40, Figure 41, Figure 42, Figure 43, Figure 44
Figure 36. The triple curve in "V", concentrated comparison of lines 4, 6 and 12 of Table 7.
Figure 37. Graphics related 10946 resonances for 36 cases of mutations associated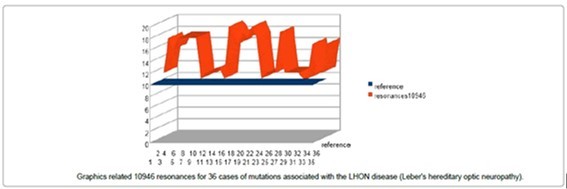
Figure 38. In 622 Germline mutations, we correlate predictive method and experimentaly
Figure 39. Predicting individual codons mutability potential in TP53 tumor suppressor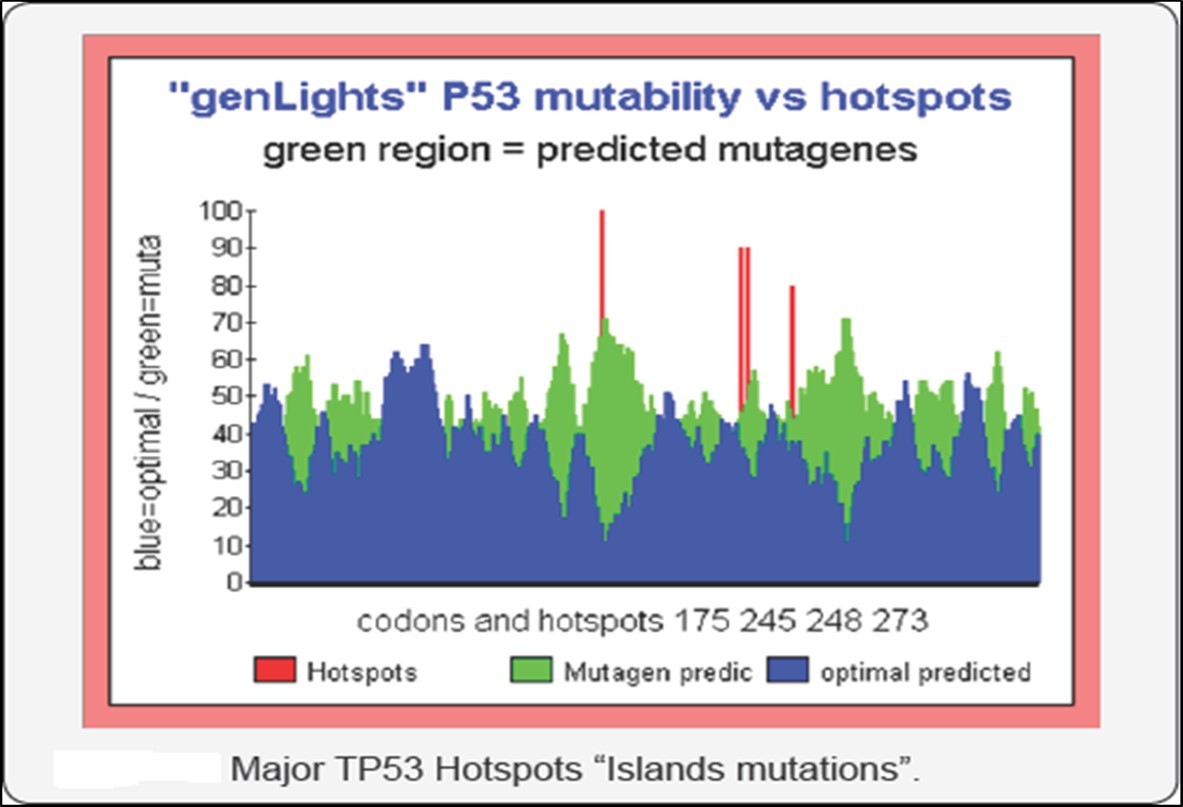
Figure 40. « physical harmonic resonance » analogy scenarii (thanks picture from Dr Robert Friedman)
Figure 41. Each DNA helix rises about 34 angstroms during each complete turn
Figure 42. Evidence of a standing wave of 34 Giga Hertz in a modelized DNA molecule using biomaterials (source publication 47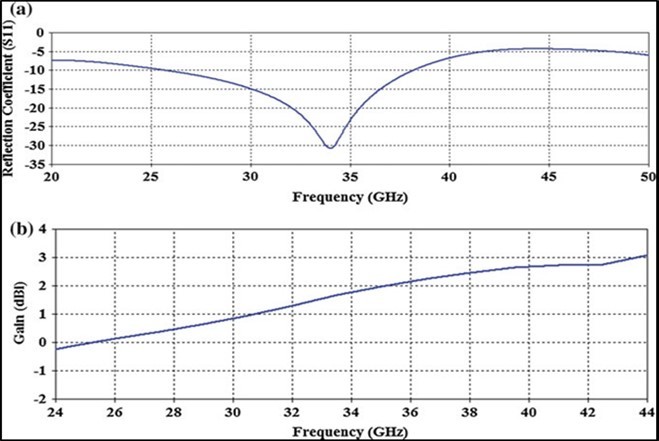
Figure 43. Fibonacci paterns emerging from plant Agave-victoriae-reginae
Figure 44. The Fractal "Dragon Curve" manage billions populations of triplets codons in the whole Human Genome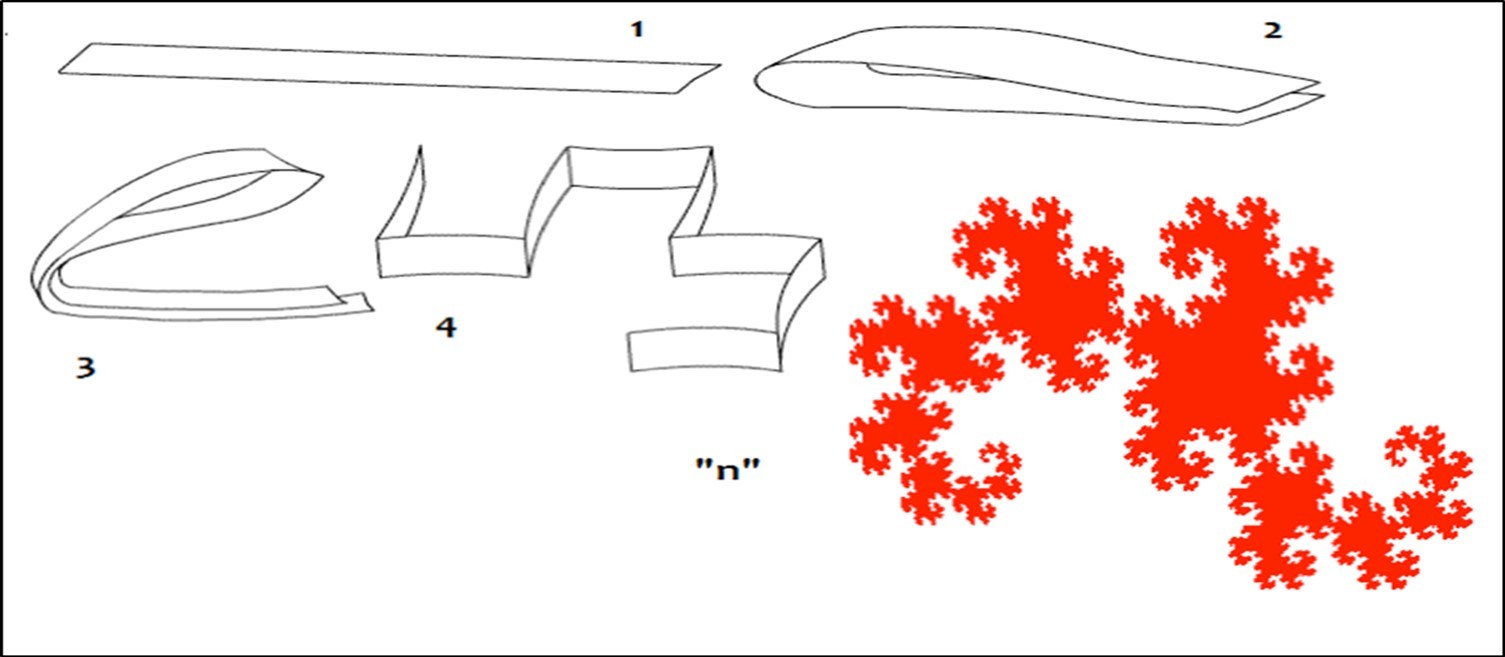
| 11 Embedded levels | Floor/FALSE/0 states:Average Floor % | Ceiling/TRUE/1 states:Average Ceiling % | Level Vote Decision |
| First Level1024 times 1000bases | 468floor level 31% | 409ceiling level 59% | Floor=FALSE |
| Second Level512 times 2000bases | 239floor level 30% | 201ceiling level 60% | Floor=FALSE |
| Third Level256 times 4000bases | 123floor level 29% | 98ceiling level 60% | Floor=FALSE |
| Fourth Level128 times 8000bases | 65floor level 29% | 47ceiling level 61% | Floor=FALSE |
| Fifth Level64 times 16000bases | 34floor level 29% | 24ceiling level 61% | Floor=FALSE |
| Sixth Level32 times 32000bases | 19floor level 28% | 11ceiling level 61% | Floor=FALSE |
| Seventh Level16 times 64000bases | 8 (*)floor level 28% | 8 (*)ceiling level 61% | UNDEFINED (*) |
| Eighth Level8 times 128000bases | 7floor level 27% | 1ceiling level 60% | Floor=FALSE |
| Ninth Level4 times 256000bases | 3floor level 27% | 1ceiling level 61% | Floor=FALSE |
| Tenth Level2 times 512000bases | 2floor level 27% | 0ceiling level 0% | Floor=FALSE |
| Final Eleven Level1 time 1024000bases | 1floor level 27% | 0ceiling level 0% | Floor=FALSE |
| Average atomic mass decreased by 0.0001 | Real average atomic mass | Average atomic mass increased by 0.0001 | |
| Inorganic astrobiologic isotopes | 18439 | 20536 | 15109 |
| Stellar Inorganic compounds | 17124 | 17067 | 13819 |
| 10000 organic compounds | 12040 | 12218 | 13662 |
| Inorganic astrobiologic isotopes | 9721 | 8312 | 10212 |
| Organiques stellaires | 12273 | 9836 | 12319 |
| 5000 combinations with parity N + H | 13922 | 5879 | 17813 |
| 153 combinations extracted from Beilstein | 22179 | 5865 | 13847 |
| 11 “citric metabolic control” compounds | 21669 | 5428 | 13259 |
| 17 common organic compounds | 8044 | 1320 | 7679 |
| 25 amino acids + UTCAG nucleotids | 16515 | 3500 | 17371 |
| Same without sulfur amino acids Cys and Met | 16646 | 1465 | 17900 |
| 18 amino acids alone (without cys and met) | 14967 | 1238 | 16758 |
| Average atomic mass decreased by 0.0001 | Real average atomic mass | Average atomic mass increased by 0.0001 | |
| 18 amino acids alone, without cys and met (line 12) | 14967 | 1238 | 16758 |
| 5000 combinations with parity N + H (line 6) | 13922 | 5879 | 17813 |
| Inorganic astrobiologic isotopes (line 4) | 9721 | 8312 | 10212 |
Cancer TP53 Mutations Prediction
In (Perez 2018b), we show how MASTER CODE could predict Mutations in the TP53 gene. These mutations are encountered in about one in every two cases of cancer. The locations and frequencies of these mutations are well known and listed. It is therefore on these mutations of TP53 that we validate here a theoretical method of prediction of the mutagenic regions of TP53. This method uses the Master Code of Biology, revealing a coupling and unification between the Genomics and Proteomics codes for any DNA sequence analyzed. The “score” of these couplings highlights the functional regions of genes, proteins, chromosomes and genomes. Of the 393 codons of TP53, and for the 61 possible values of these codons authorized by the genetic code (i.e., 393x61 genes simulated), we prioritize the corresponding Master Code scores. Codons with scores close to 1 correspond to conserved regions whereas codons with scores close to 61 reveal highly mutagenic regions. Our method is then validated and correlated with the real mutations observed experimentally on hundreds of cases. We then analyze the potential of this method in the context of future quantum computers.
Conclusions
We will conclude here by interesting the different following themes that emerge from the reflexions raised by these 6 codes:
The Diversity
The law of numerical projection atomic mass code (-I-) behaves like a kind of ideal law, objective in the sense defined by Jacques Monod 13. Because it is optimal for the average atomic masses of bioatoms, it is inaccessible in the real universe which is only individual isotopes. Thus this law, if the nature of the living "seeks" to approach it in search of better equilibrium and stability, will compel to seek more DIVERSITY. Diversity of isotopes of the same atom, diversity of isotope changes over time. We can make an analogy with the electron layers of atoms: they are only densities of probability, an individual electron only reaching by chance this ideal value. Here it is the same: this optimal ideal code for the average atomic mass that never exists in the real world of isotopes will force them to diversify and self-organize for more stability ... as if the forced plan of evolution towards more complexity (molecules, RNA, DNA, amino acids) was already a necessity, an implication, in the sense dear to Nobel prizewinner Jacques Monod.
The Unification
Contrary to the appearances established by the "Central Dogma of Biology", there would not exist 3 distinct and heterogeneous types of genetic information (DNA, RNA and amino acid sequence) but only one "meta-information" such that, to any double-stranded DNA sequence (coding or junk-DNA), we can ALWAYS, after projection according to the atomic mass code (-I-), associate 3 unified images named respectively: genomic signature, proteomic signature and RNA signature, such than:
-the RNA image is always empty and zero.
-the 2 genomic and proteomic images are always almost identical and correlated more than 99%.
Everything happens as if an inaccessible unique and virtual information, "implied" in the sense of the physicist David Bohm 14, "unfolded" materially according to the 3 physical media or respective languages of the DNA , RNA and amino acids, so that one of the 3 images is "empty" (RNA) and the other 2 similar and equivalent (DNA and amino acids).
The Fractal Textures
A fine analysis of the "texture" of these curves Genomics and Proteomics will now reveal a strange phenomenon: a curious roughness "sawtooth" (Figure 7) characterizes most of these images. It is a kind of search for the "derivative of order 1", that is to say the "slope" between 2 successive points. We then note that these slopes are mainly of the same direction: always increasing or always decreasing.
After successive applications of the first and second codes (-I- and -II-) to any genomic double stranded DNA sequence, the texture analysis (roughness) of the genomic and proteomic images highlights: Figure 8
-a modulation in the form of a ANALOGIC distribution (Figure 9) for the Genomic code.
-a modulation in the form of a 0/1 BINARY distribution (Figure 10) for the Proteomic code.
Everything happens as if: two different languages (genomics and proteomics) produced two very similar images but radically different roughnesses: the first (genomics) would be analogous to an amplitude modulation, while the second one (proteomics) would be analogous to a frequency modulation (to use the analogy of radio sounds and waves). We can even say that the second would correspond to a coding of these sounds in binary form as we find them in compact disk or in the current laser based digital coding of information.
A second analogy: suppose that a same sequence of information is coded and transmitted according to two radically different languages (for example in French and in English), These 2 encodings will produce the same "meaning", the same "comprehension", the same mental representation of the transmitted message. However, the "roughness", (% of vowels of each of the 2 languages for example) will be radically different.
Numbers and Codes of Life
One of the most fascinating questions of mathematics is the relation between these two universes of numbers, so different as real numbers and integers. One models the universes of the continuum while the other represents the discrete, quantum universes. .. Here, with these 6 successive nested codes we went from a continuous universe of real numbers of atomic masses to the discrete universe of integers encoding Pi-masses. Then, with the Master code we continue in a universe of integers producing these 2D Genomics and Proteomics images. Then, with the biobits of the binary logic code of the proteomic textures, we comets the universe of the integers in a binary universe with only 2 states, those of the logic. Then, with discrete waves, standing waves and resonances, it is again in a continuous world that we come back: the wave universe ... Thus, over these 6 codes, the 3 digital worlds of the continuous, the discrete and binary have always been ubiquitous. We think that biology and genetics are inseparable from the world of numbers!
Geometry and Codes of Life
Phi = 1.618033 ... the "golden number" is omnipresent in Nature, often in the form of proportions according to Fibonacci numbers (1 1 2 3 5 8 13 21 34 55 89 ...), as in the pinecone or pineapple 15.
From the 80s, in our research in Artificial Intelligence, we demonstrated with our neural model “Fractal Chaos” a natural link between fractals 16 and the golden ratio 17, 18, 19.
In this paper, the analysis of the genetic information (proteomic image) of the entire human genome highlights a binary modulation (Figure 15 and Figure 16) throughout the genome.This binary modulation is distributed statistically around 2 binary attractors whose ratio is equal to TWO and whose respective values are phi (level "high" = 1) and phi / 2 (level "low" = 0) With phi = 0.618 ... = 1 / Phi the Golden ratio (Phi = 1.618. ..).
More generally, since 1991, we have described the presence of Phi the Golden ratio and that of Fibonacci numbers in DNA, genes, chromosomes and genomes in many forms 20, 21, 22, 23, 24, 25, 26, 27, 28.
Finally, Golden ratio and Fibonacci numbers are an evidence in DNA numerical structure, see by example these « 34 » scale invariance fractal occurrences of Fibonacci number 34 :
Recently published, in 29, in « DNA as an Electromagnetic Fractal Cavity Resonator: Its Universal Sensing and Fractal Antenna Behavior », P. Singh et al. Simulated the DNA geometry using biomaterials, they write : « then, they demonstate that 3D-A-DNA structure behaves as a fractal antenna, which can interact with the electromagnetic fields over a wide range of frequencies. Using the lattice details of human DNA, we have modeled radiation of DNA as a helical antenna. The DNA structure resonates with the electromagnetic waves at 34 GHz ».
Thus, the reader can now reflect on this strange trilogy:
Each DNA helix rises about 34 angstroms during each complete turn.
We have also demonstrated that the chromosomes 4 of Sapiens and Neanderthal have a standing wave of 34 nucleotides 30.
Singh et al. demonstrate in 2017 that a standingwave of 34 Giga Hertz would organize DNA molecules 29...:
Finally, DNA, mathematics 31 and stationary waves 32, 33 form a rich intersection, which many publications affirm.
Speculations
Thoughts on this article about the MATTER, WAVE and INFORMATION triptych
3 stages of speculations about the MATTER INFORMATION WAVE triptych:
1 / in the last century, a central subject of reflection was that of the WAVE PARTICLE Duality (Albert Einstein, louis de Broglie).
The measurement (INFORMATION) of the particle excludes that of the wave associated with it and vice versa.
One cannot thus obtain simultaneously the 2 states WAVE and MATTER (particle) and measure, therefore INFORMATION.
2 / in this article, we realize the union of the 3 concepts in the direction (MATTER + INFORMATION) ==> WAVE.
Either atomic masses + DNA sequence ==> Standing waves.
We can prove it, on the one hand by calculating the cumulative mass of the millions of atoms constituting a chromosome according to its sequence, and, on the other hand, by showing the standing wave associated with it.
3 / curiously, in the memory of water applied to DNA (Montagnier, 2017), it is the INVERSE process of (2) which would occur:
(WAVE + PCR) ==> DNA
So THE WAVE that implicitly contains the INFORMATION would return the MATTER (DNA) through the PCR operator.
Here also, we realize the union of the 3 concepts but in the direction (WAVE containing the INFORMATION) ==> MATTER.
But a 4th "actor" has just been added here: the central role played by WATER H2O ...
Perspectives as an Extension of the Preface by Luc Montagnier Six Others Codes of Life
A question which intrigues Luc Montagnier these days concerns that of the differentiation of cells from this initial cell. He also insists on the 2 ways of perceiving the genome: continuous sequence and structured in codons. Finally, he wonders about the central role that fractal growth and waves could play in this process of emergence of a life.
This perspective forces me to revisit this article (Perez, 2010) popularized in
(https://www.sacred-geometry.es/?q=en/content/phi-and-music-dna)
In which I demonstrated how a billion codon triplets of the human genome, when they were re organized through the kaleidoscope of the 64 values of the genetic code divided into 6 successive partitions following the famous fractal curve of the dragon, alternately brought out 2 fractal attractors 1 and (3-Phi) / 2. The first following TC / AG type partitions, and the second, following TA / CG partitions, precisely those that Luc Montagnier mentioned as obeying Fibonacci numbers in the evolution of SARS-CoV2 variants.
From there to sketching out a line of research where cell differentiation would obey an evolution via this Dragon curve, there was only one step: indeed, these differentiated cells do indeed produce living matter, therefore proteins, therefore. a logic of triplet codons.
The fascinating fact is that, what we call "PROTEOMICS IMAGE" in this article, corresponds to a potential proteome of 3 times a billion codons, therefore potential amino acids (3 times due to the 3 possible reading frames of the codons of DNA). This potential proteome, a tiny part of which is materialized in proteins, constitutes a sort of "infinite reserve" of future pending proteins.
Luc Montagnier's "bet" at the end of his preface can now be deciphered by the extremely rare reader who is too curious about this article.
Are FRACTALS and WAVES already engraved in our genome?
Would there exist 3 nested - entangled - codes in the monotonic sequence of the human genome, "eternal garland" according to Douglas Hofstadter in "Godel, Escher Bach"?
The answer is yes"!
The first of the 3 CODES is linear, analog, it is the simple sequence of the genome. In this article, we can see it in the thin blue curve of Figure 15 and Figure 16.
The second CODE is BINARY and FRACTAL, we can see it in the red binary curve of these same Figure 15 and Figure 16. Why is it fractal? Because, as in the famous Dragon curve (table of the universal genetic code) in (Perez, 2010), it results from a choice, from a bifurcation, with each new nucleotide in the genome, it is this new nucleotide which will produce the choice, the bifurcation, in this dichotomy of the codons of the genome in 2 families, that of the attractor "1" (codons TC = codons AG), and that of the attractor "(3-Phi) / 2" (TA / CG codon ratio).
The third CODE is WAVING, it comes from meta-codons of amplitude 4, 5, 6 .... n nucleotides described in our method of calculating waves ... It is a medium distance order, unifying each chromosome by a standing wave ...
Acknowledgements
Thanks for fructuous discussions about this article to, Robert Friedman M D. (author of "Nature's secret nutrient, golden ratio biomimicry, for PEAK health, performance and longevity), Valère Lounnas, (Free lance researcher at CMBI European Molecular Biology Laboratory (EMBL) Heidelberg) for a nice « MASTER CODE » results reproduction in https://www.francesoir.fr/histoire-du-covid-19-chapitre-9-partie-2, Ethirajan Govindarajan (adjunct Professor, Department of Cybernetics, School of Computer Science, University of Petroleum and Energy Studies, Dehradun, Uttarakhand, India, Director, PRC Global Technologies Inc., Ontario, Canada, President, Pentagram Research Centre Pvt. Ltd., Hyderabad, India), Mrs Marie-Laure Becquelin for discussions on MATTER, WAVES and INFORMATION concept (paraisomiamibeach.com)
and http://science-anthology.com), dr Marco Paya Torres MD Alicante, dr Richard M Fleming PhD, MD, JD ( https://www.flemingmethod.com/) and Xavier Azalbert, Director FRANCE-SOIR newspaper ( https://www.francesoir.fr/ info-en-direct ).
We thanks also Megawaty Tan (A private researcher based in South Sumatera, Indonesia), Sami MacKenzie-Kerr private researcher in Bali ("The Matrix") https://matrix.fandom.com/wiki/The_Matrix/Crew mathematician Pr. Diego Lucio Rapoport (Argentina), Volkmar Weiss (Dr. rer. nat. habil. Dr. phil. Habil. Leipzig, Germa ny), and Dr Xavier Bezançon (Paris) for various discussions, and Professor Sergey V. Petoukhov (Dr. Phys.-Math. Sci, Grand Ph.D. Full Professor, Laureate of the State prize of the USSR), which reproduced then published some results at whole human genome scale.
Finally, this work is the result of multiple exchanges and advice, since 1994, for which I must thank Professor Luc Montagnier (Nobel prizewinner for his discovery of HIV, Fondation Luc Montagnier Quai Gustave-Ador 62 1207 Geneva, Switzerland).