Abstract
Perkinsus marinus is an intracellular parasitic protozoan that is responsible for serious disease epizootics in marine bivalve mollusks worldwide. Despite all available information on P. marinus genomics, more baseline data is required at the proteomic level. Our aim was to study the proteome profile of in vitro cultured P. marinus isolated from oysters Crassostrea spp. using a label-free shotgun UDMSE approach. A total of 4073 non-redundant proteins were identified across three biological replicates with stringent identification. Proteins specifically related to adaptive survival, cell recognition, antioxidants, regulation of apoptosis and others were detected. Important virulence factors of P. marinus were identified including serine protease and iron-dependent superoxide dismutase. Other proteins with involvement in several pathogens invasion strategies were rhoptries, serine-threonine kinases, and protein phosphatases. Interestingly, peptides corresponding to retroviruses polyproteins were identified in all replicates. The interactomic analysis of P. marinus proteins demonstrated extensive clusters network related to biological processes. In conclusion, we provide the first comprehensive proteomic profile of P. marinus that can be useful for further investigations on Perkinsus biology and virulence mechanisms.
Author Contributions
Academic Editor: Leonid Tarassishin, Department of Biological Sciences.St. John's University.United States.
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2017 Márcia P. Leibowitz et al
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Introduction
Marine bivalve molluscs production worldwide is being affected by disease epizootics caused by intracellular protozoan parasites of the genus Perkinsus. Numerous species have been described but only two species namely Perkinsusmarinus and Perkinsusolseni require notification to the World Organization for Animal Health 1. P. marinus has been associated with significant mortalities of the eastern oyster Crassostrea virginica but other species such as the pacific oyster Crassostrea gigas and the mangrove oyster Crassostrea rhizophorae are also susceptible 2.
The parasite belongs to a new independent phylum Perkinsozoa that is positioned between the phyla Apicomplexa and Dinoflagellata 3. Species of Perkinsus are believed to be transmitted directly from host to host during the feeding process 4. Different developmental stages are observed that are all believed to be infective 2. No effective therapies have been developed to date.
A considerable amount of genomic information is availablefor P. marinus. In contrast, few studies available on comparisons of proteomic profiles of different Perkinsus species and on proteome variability of P. olseni resulted in low throughputs: 28 and 19 annotated proteins, respectively 5, 6. More proteomic baseline data is required for a better understanding of P. marinus biology, including virulence mechanisms. Proteome techniques nowadays allow us to observe whole cellular events by directly visualizing the proteins being expressed. Neither genome nor transcriptome investigations would allow the analysis of such complex parasitic responses 7.
In the present study, we analyzed the proteomic profile of in vitro cultures of P. marinus by a high throughput label-free shotgun UDMSE approach using nano ultra-performance liquid chromatography mass spectrometry (nanoUPLC-MS).
Material and Methods
P. marinus cultures were established from infected gills and mantle tissues of native oysters of the genus Crassostrea obtained from São Francisco do Sul, State of Santa Catarina, Brazil, following methods developed for Perkinsus spp. isolation 8. Clonal cultures of trophozoites were established through isolation of individual cells by a standard limiting dilution method until it reached exponential phases of growth in DMEM/F-3 medium 9. Parasite densities were estimated by counting with a hemocytometer.
Proteomic analysis was conducted using P. marinus (1,7 x 108 cells) with three biological replicates. Briefly, P. marinus cells were centrifuged (1000 g for 10 min at 10 °C) and proteins were extracted by suspending pelletedcells in lysis buffer (42 % urea, 15 % thiourea, 4 % CHAPS, 12.5 mM Tris-HCl pH 7.5 and 1.5 % DTT). A protease inhibitor mix at 1 % (GE Healthcare, UK) was also added. Samples were then centrifuged (22000 g) at 4 °C for 40 min and washed four times in 50 mM NH4HCO3. Proteins were quantified using Qubit protein assay kit (Thermo Fisher, UK).
Samples were submitted to tryptic digestion using 50mM NH4HCO3 and 0.2% RapiGest SF (Waters) at 80 °C for 15 min, following treatment with 100 mM DTT at 60 °C for 30 min and then carboxamidomethylated in 300 mM of Iodoacetamine at room temperature for 30 min. Next, trypsin (Promega, USA) were added and incubated at 37 °C for 16 hours. Finally, 5% TFA was added at 37 °C for 90 min and samples were then centrifuged (22000 g) at 6 °C for 30 min. Supernatants were then transferred/ to the Total Recovery Vials (Waters, USA) and subjected to nanoUPLC-MS analysis using a Synapt G2Si mass spectrometer.
Qualitative bidimensional nanoUPLC (multiplexed DIA - data-independent acquisition) analysis were conducted using both a 1-h reverse-phase gradient from 7 % to 40 % (v/v) acetonitrile (0.1 % v/v formic acid) and a 450 nL.min-1 nanoACQUITY UPLC 2D Technology system. MS analysis of tryptic peptides was performed using a mass spectrometer equipped with a T-Wave-IMS device in MSE and UDMSE modes 10. MSE and UDMSE raw data generated for each replicate were submitted to ProteinLynx Global Server software (PLGS) version 3.0.2 (Waters), with the following settings: maximum of 1 missed cleavage by trypsin, a fixed modification: carbamidomethyl (cysteine) and variable modification: oxidation (M). Mass spectra were searched against P. marinus protein database UniProt (Proteome ID UP000007800; 23,114 proteins). Protein identification was considered valid only when the following criteria were met: auto curate value Green (representing 99% of spectrum confident), default maximum false discovery rate (FDR) of 4 % and proteins with at least two distinct peptides. FDR were determined by searching against the reversed-sequence decoy database version of P. marinus protein sequences. For quantitative analysis, the dynamic range of protein abundance was calculated using the average of "MatchedProductIntenSum" column of PLGS data of all replicates. The data was decreasingly ordered and plotted using R software 11 (Figure 3). A protein-protein interaction network was built using the algorithm STRING 12. STRING data setting adjusted parameters were active interaction sources to experiments, gene fusion, databases, co-ocurrence and co-expression; and the minimum required interaction score to custom value of 0.980. Proteins present in all replicates with at most a unique peptide were included as query in STRING web server (n = 881).
Results
A total of 4073 non redundant proteins were identified across the replicates and a combined total of 2810 proteins were present in at least two of the biological replicates (Figure 1). The calculated FDR was ~1 % when detection was set at agreement of all replicates. The complete list of identified proteins can be found in the Supplementary Table S1. The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE 13 partner repository with the dataset identifier PXD003727.
Figure 1. Venn diagram showing the numbers of unique and overlapping proteins identified between all biological replicates (R1, R2, and R3). 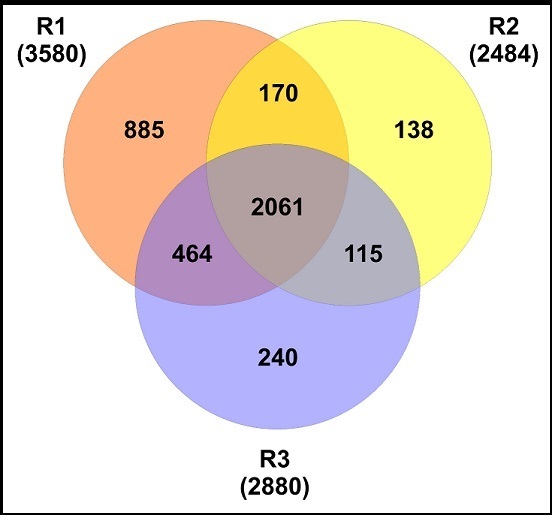
The identified proteins were divided into 14 categories, according to Gene Ontology (GO) classification. Most of the identified proteins (82.09 %) were involved in metabolic, cellular and single-organisms processes. Other identified proteins were involved in response to stimulus, localization, biological regulations, cellular component organization or biogenesis, signaling, detoxification, developmental process, biological adhesion and less than only 1 % were involved in multicellular, organismal process and locomotion (Figure 2). The predicted proteins were blasted against Uniprot/TrEmbl local database with e-value of 1e-06, percent of identity of 90, and maximum number alignments of 20. The GO function was mapped using a local database. Briefly, 907 (~26 %) proteins had unknown functions, whereas 4860 GO terms were assigned on Level 2 for Biological Process to the other 3166 proteins. The dynamic range (~3 logs) of quantified proteins of P. marinus was analyzed aiming to generate a global view of their relative abundance distribution. In our study, it was observed a high estimated abundance of proteins that were mostly related to biological processes and virulence in P. marinus (Figure 3).
Figure 2. Classification of all detected proteins of in vitro cultures of P. marinus, using GO annotation. The distribution was made based on biological processes.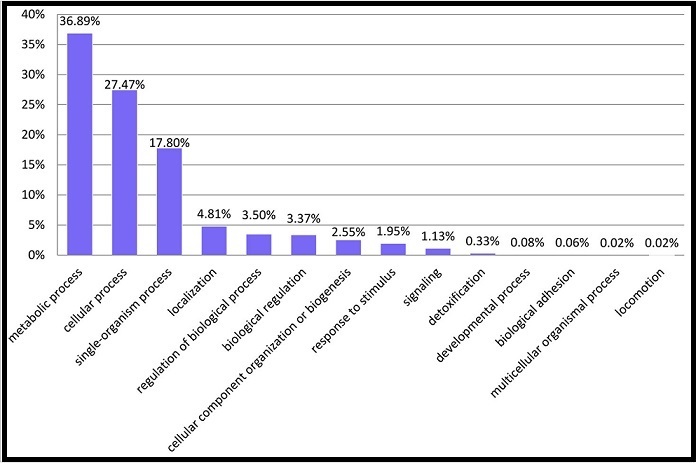
Figure 3. The dynamic range of the method based on protein abundance estimates, data points derived from the LC-UDMSE analysis. Y-axis shows log10 of Hi-3 matched peptides abundance for each protein. Yellow square shows the proteins cited along this work.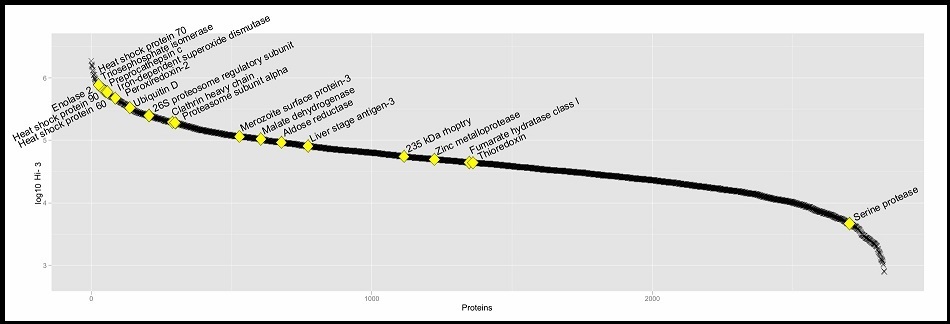
Results from the interactomic analyses revealed that the greatest number of interactions were verified in the proteins related to ubiquitin-proteasome complex (cluster with 41 proteins), ribosomal proteins (41 proteins), iron-sulfur fumarate hydratase (20 proteins), TCP-1 (15 proteins), ATP synthase (17 proteins), pyruvate ferredoxin (7 proteins), triose phosphate isomerase/phosphoglucose isomerase/enolase (14 proteins) and clathrin interactome (15 proteins) (Figure 4).
Figure 4. Protein-protein interaction networks of Perkinsus marinus proteome. STRING algorithm was used to build an interaction map among the proteins identified in all three biological replicates. Thicker lines denote interactions with score ≥ 0.980. (1) Ubiquitin-proteasome complex. (2) Ribosomal protein cluster. (3) Iron-sulfur/fumarate hydratase cluster. (4) T-complex protein-1 (chaperonin) cluster. (5) ATP synthase network. (6) Pyruvate ferredoxin. (7) Triosephosphate isomerase/phosphoglucose isomerase/enolase cluster and (8) Clathrin interactome.
Discussion
The proteome of in vitro cultured P. marinus was analyzed. In this study, we provided the first broad-based proteomic view into the basic biology and cellular metabolism of P.marinus.
Differences were observed in the number of identified proteins between biological replicates. Species of Perkinsus under culture conditions exhibits morphologically distinct life stages 8 making it quite difficult to produce homogeneous cultures of P. marinus. These differences would probably have an effect on the nature of the protein being expressed by each biological replicate.
For proteome dataset analyses, particular attention was given to proteins that are known to represent virulence factors in P. marinus and in other pathogens, including closely related apicomplexans. Several heat shock proteins were detected in all replicates mostly from the 70 kDa family that are the most common expressed heat shock proteins in response to stress 14, 15. Heat shock proteins are necessary for P. marinus adaptive survival repertoire 16, 17. Antioxidant proteins including peroxiredoxin-2 and thioredoxin were detected in all triplicates, as well as proteins that are known to have immunosuppressive action, such as cyclophilins 17. Proteins that are involved in the regulation of apoptosis including TP53-regulated inhibitor of apoptosis, liver stage antigen 3, pterin-4a-carbinolamine dehydratase, ubiquitin, polyubiquitin and adenylate kinase B were identified in at least two replicates 15, 17.
Serine proteases and iron-containing superoxide dismutase that are suggested to be virulence determinants of P. marinus were detected in at least two replicates 18, 19. Proteases are known to play important roles in P. marinus disease pathogenesis causing cellular and tissue damage 17, 18. Other identified proteases of great importance in virulence included: serine/threonine kinases, cathepsin b, c, L and z, preprocathepsin c precursor, Thiolproteinases, aspartyl aminopeptidase, 26S proteases regulatory subunit, proteasomes, m1 zinc metalloprotease, intracellular alkaline proteases and cysteine protease 15, 17.
Additional proteins that are important for the pathogenesis of P. marinus and a variety of bacterial and parasitic organisms were detected in at least two replicates, including: aldose reductase 20, betaine aldehyde dehydrogenase 21, helicases 15, 22, malate dehydrogenase 6, and aspartyl aminopeptidase 23.
Rhoptries are club-shaped secretory organelles that discharge their contents during infection. Rhoptry proteins were detected in all replicates. P. marinus is well known to infect oyster hemocytes residing inside phagosome-like vesicles where they remain viable and multiply. Rhoptries are considered to be key mediators of virulence by enabling many parasites, including apicomplexans to invade hosts erythrocytes 24. Rhoptries contains a number of novel proteins (ROP) including serine-threonine kinases and protein phosphatases that were detected in all replicates and are also believed to be parasitic virulence determinants 24. Furthermore, the merozoite surface protein 3 was detected in all replicates. This protein is reported to be important parasitic surface antigens as well as virulence factors 25.
One remarkable finding was that peptides corresponding to retroviruses polyproteins were detected. An analysis of the P. marinus genome reveals that there are three putative endogenous retroviruses present and our findings shows that these sequences are being transcribed and translated. The presence of a virus and/or retrotransposon elements in the P. marinus genome based on genomic annotation together with EST data was previously reported 15. The implications of the presence of these viruses are still unknown; however, they might have an impact in Perkinsus survival and possibly even its pathogenicity.
A protein-protein interaction network was analyzed to better understand the biological outcome of the detected proteins of P. marinus and a great number of interactions were observed. A prominent feature of the life cycle of Perkinsus spp. is the numerous morphological changes they undergo during development in the oysters hosts, which must involve extensive and carefully controlled proteolytic activity. There are several reports that have confirmed the role of proteasomes in parasite differentiation and proliferation processes, which are key steps in pathogen colonization 26. A great variety of proteasome inhibitors have been studied as molecular targets for treatment development of many parasitic diseases by selectively killing or disrupting parasite multiplication 26, 27. It would be interesting to examine P. marinus proteasome as potential drug targets for mollusc diseases. Abundant interactions for ribosomal proteins might be suggestive of the rapid and extensive protein translation that accompanies parasite differentiation and multiplication following host-cell invasion 15. The iron-sulfur cluster fumarase activity has been shown to be essential for protozoan parasites such as Trypanossoma cruzi since its mitochondrial isoform is part of the tricarboxylic acid cycle and as such being central to aerobic respiration 28. The TCP1 complex belongs to the HSP60 family protein that was detected in P. marinus from our study. This protein is involved in folding and assembly of wide range of cytosolic proteins after stress related denaturation being critical for maintaining the integrity of cellular proteins 15. In most organisms, ATP synthase taps the energy stored in the proton gradient generated by the respiratory complexes to synthesize ATP from ADP and phosphate (Pi). However, it has been suggested that in blood-stage trypanosomes that has no mitochondrial respiratory enzymes, this enzyme might operate in reverse, as an ATPase hydrolyzing ATP into ADP and Pi and pumping protons into the intermembrane space. This reverse reaction would be important to maintain the membrane potential during anoxia 29. Since P. marinus primarily infects host hemocytes, it is interesting to further examine Perkinsus ATP synthase mechanisms. It has been reported that P. marinus possesses genes for a plant-type ferredoxin system that possibly encodes plastid-targeting signals 30. In addition to ferredoxins, triose phosphate proteins that are also predicted to target plastids were detected in our study, however its implications are unknown. Clathrin-mediated trafficking is known to be responsible for endocytosis and post-Golgi transport in trypanosomes. It also represents an important interface with the host, in addition to play multiple roles in immune evasion and host cell invasion that are vital for effective infection and persistence 31.
In conclusion, this is the first comprehensive identification of P. marinus proteins by a label-free shotgun proteomic approach. These results might serve as a valuable resource for future investigations involving comparative proteomics, potential drug targets, mechanisms of adaptation under stress-related conditions, as well as host-parasite interactions.
Supplementary table
Acknowledgements
This study was supported by the Ministry of Agriculture, Livestock and Food Supply (MAPA) and the National Center for Animal Information (INCT)/CNPq/UFMG. We thank FAPEMIG, CAPES, and CNPq.