Abstract
Cancer is the leading cause of death worldwide, and there is a constant need for new treatment strategies. Sesquiterpene lactones containing a 3-methylenedihydrofuran-2(3H)-one (or α-methylene-γ-lactone) moiety, for example damsin (1), are Michael acceptors that affect biological processes such as cell proliferation, death/apoptosis, and cell migration, by interfering with cell signalling pathways. Although the reactivity of the α-methylene-γ-lactone moiety is important for these effects, the Michael addition is reversible and it can be assumed that also other parts of the molecules will moderate any given biological activity. In this investigation, the cytotoxicity of 23 -methylene--lactones towards normal breast epithelial MCF-10A cells as well as breast cancer JIMT-1 cells is compared. Most of the investigated compounds are semisynthetic derivatives prepared by the condensation of the natural product damsin (1) with aldehydes. The two cell lines were treated with various concentrations of the compounds in dose response assays, and the 50 % inhibitory concentration (IC50) was determined from dose response curves. The IC50 values were found to depend strongly on the overall structure. The ratio between the IC50 values for MCF-10A and JIMT-1 cells, as a measure for the selectivity of a compound to kill cancer cells, was calculated, and found to vary between just over 1 to more than 10. The most potent derivatives formed from the condensation of 1 with aromatic aldehydes towards JIMT-1 cells are 3a and 3i, both with ratios between the IC50 values for MCF-10A and JIMT-1 cells close to 5. Also some aldol condensation products with acyclic aldehydes, i.e. 3r and 3u, were equally potent, and the latter showed the highest selectivity (ratio > 10). Structure-activity relationships that may explain the observed differences in potency and selectivity are discussed.
Author Contributions
Academic Editor: Sahar Ahmed, University of Alberta, Canada.
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2019 Maribel Lozano, et al.
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
No author has any associations that may represent a potential conflict of interest.
Citation:
Introduction
The incidence of cancer is increasing, and different strategies for controlling the disease are developed. The pool of secondary plant metabolites has always been an important provider of low-molecular anti-cancer drugs, and is expected to be so also in the future. As our understanding about the molecular mechanisms of cancer development and progression, as well as the development of treatment resistance, has increased, our ability to design new anti-cancer drugs has improved.1,2 One of the cellular molecular pathways that is important for chemoresistance and metastasis in many cancer cells is the NF-κB pathway, where constitutive activation, or over-expression, is part of the tumour aggressiveness.3 Terpenes containing an α-methylene-γ-lactone moiety are natural products that have been shown to possess promising anti-cancer effects, interfering with biological processes such as cell signalling, proliferation, death/apoptosis, and migration.4,5 The significant cytotoxic activity of such terpenes is linked to the α-methylene-β-lactone moiety, which via a Michael addition can alkylate nucleophilic residues (primarily free thiols of cysteines) in proteins. A target suggested for such an interaction is the protein p65 in the NF-κB pathway.6
In general, electrophilic compounds are considered potentially toxic and have been rejected by the pharmaceutical industry in their search for novel drug candidates.7,8 However, 39 electrophilic drugs, mainly used in oncological therapies, have been approved by the US Food and Drug Administration.8 These will display more or less off-target toxicity, including immunogenic responses.8 The efficiency and selectivity of an electrophilic compound will besides the reactivity of the electrophilic moiety depend on the affinity to the target through non-covalent interactions. Michael acceptors will react with nucleophiles via an addition reaction, not a substitution, in a reaction that is reversible, and will bind tighter to nucleophiles that present a more favourable molecular environment. However, the identification of specific targets and structural features of non-covalent interactions of Michael acceptors has been elusive and complex to study.
Several studies of structure-activity relationships (SARs) of natural terpenoid Michael acceptors have shown that guaianolide and pseudoguaianolide sesquiterpenes with a α-methylene-γ-lactone moiety possess the most potent anti-cancer and cytotoxic activities.9,10 Although such sesquiterpenoids are considered to interact with multiple targets in cancer cells, they have been shown to inhibit transcription factors such as NF-κB, STAT3, and AP-1, up- or downregulate the protein kinases MAPK and JNK, and increase the expression of the p53 protein.11 Besides that, they have been shown to disrupt the redox balance and induce an oxidative stress in cancer cells.12
In the present study we compare the cytotoxicity of 23 α-methylene-γ-lactones based on the natural product damsin (1) (see Figure 1), in normal breast epithelial MCF-10A cells and breast cancer JIMT-1 cells. 1 is a pseudoguainolide sesquiterpene with a moderate inhibitory activity on NF-κB,13 available from the plant Ambrosia arborescens together with the 1-hydroxy derivative coronopilin (2). Compound 1 was used as the starting material for the semi-synthetic preparation of 21 novel analogues.
Figure 1. aThe condensations to eventually give 3l, 3m, and 3n were carried out with the MOM-protected hydroxybenzaldehydes, prepared according to Figure 2 and the Experimental section. As damsin (1) was isolated as a pure enantiomer, the absolute configuration for all compounds is as shown in Figure 1.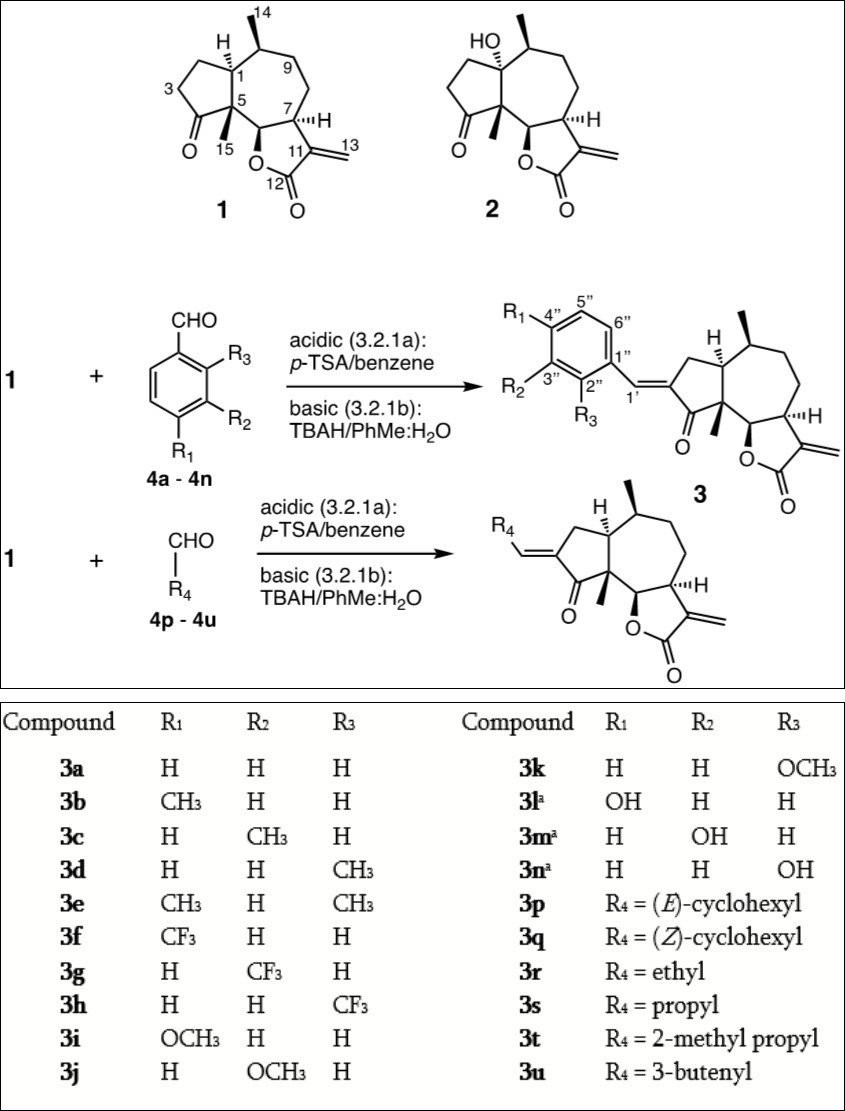
Experimental Procedure
General Chemical Procedures
Chemicals (analytical grade) were purchased from different commercial suppliers and used without further purification. Damsin (1) and coronopilin (2) (Figure 1) were isolated from A. arborescens as previously described.13 IR spectra were recorded with a Brucker Alpha FT-IR Spectrometer, and the optical rotations was measured with a Perkin-Elmer 141 polarimeter. HRMS spectra were recorded with a Waters XEVO-G2 QTOF instrument, while NMR spectra were recorded in CDCl3 using a Bruker DRX spectrometer operating at 400 MHz for 1H and at 100 MHz for 13C. Chemical shifts are given in ppm relative to the solvent signals (7.26 ppm for 1H and 77.00 ppm for 13C). All compounds described here were completely analysed by 2D NMR (COSY, HMQC, HMBC and NOESY), and as the syntheses start with the pure enantiomer damsin (1) all compounds are given with the absolute configuration in Figures 1 and 2. Chromatography was performed with 60 Å 30-75 μm silica gel, while TLC analyses were made on Silica Gel 60 F254 (Merck) plates.
1. Synthetic Procedures
1a. General Procedure for Claisen-Schmidt/aldol Condensations Under Acidic Conditions
A mixture of 1 (1 eq., 0.4027 mmol), aldehyde (1.3 eq., 0.5235 mmol), and p-TsOH (1.5 eq., 0.6040 mmol) in benzene (10 ml) was prepared in a sealed tube, and stirred at 80 °C for 19 to 66 h until the reaction was completed (monitored by TLC). The reaction was quenched by adding 2 ml of 5 % NaHCO3 followed by 15 ml brine, and the mixture was extracted with DCM (3 x 15 ml). The combined organic layers were dried with MgSO4 and concentrated in vacuo. The crude product was purified by silica gel chromatography (EtOAc:petroleum ether 1:1).
1b. General Procedure for Claisen-Schmidt/aldol Condensations Under Basic Conditions
A mixture of aldehyde (3 eq., 1.2081 mmol) and 1 (1 eq., 0.4027 mmol) in 50 % PhMe:H2O 1:1 (3 ml) was cooled in an ice-bath, and TBAH (1.2 eq., 0.4832 mmol, 1.5 M in H2O) was added dropwise. The ice-bath was removed after 10 min and the reaction was left to reach room temperature. The reaction was monitored by TLC, and after 3 to 24 h the reaction was quenched by addition of 2 ml of 5 % HCl and stirred for 15 min at r.t. The reaction mixture was diluted with 15 ml brine, and extracted with DCM (3 x 10 ml), and the combined organic layers were dried with MgSO4 and concentrated in vacuo. The crude condensation product was purified by silica gel chromatography (EtOAc:petroleum ether 1:1).
1c. General Procedure for the MOM-Protection of the Hydroxybenzaldehydes.
To a solution of hydroxybenzaldehydes 4l – 4n (284.9 mg, 2.3333 mmol) in 6 ml dry DCM was added EtN(i-Pr)2 (2 ml, 11.6665 mmol), followed by MOMBr (400 μl, 4.8999 mmol) dropwise under N2 at 0 °C, whereafter the mixture was stirred at room temperature for 6 h. The reaction was quenched with 10 ml of a saturated solution of NaHCO3 by stirring for 15 min at r.t., and the mixture was extracted with DCM (2 x 15 ml). The combined organic layers were dried with MgSO4 and concentrated in vacuo. The product was purified by chromatography on silica gel (n-heptane:DCM 1:1).
1d. General Procedure for the Deprotection of the Claisen-Schmidt Adducts with MOM-Protected Hydroxyl Groups.
To a solution of a MOM protected Claisen-Schmidt adduct (50 mg, 0.1261 mmol) in MeOH (2.6 ml), HCl conc. (22.5 μl, 0.2695 mmol, 37 %) was added slowly. The reaction mixture was heated to 40 °C for 5 h and then cooled to room temperature, whereafter the organic solvent was evaporated in vacuo. The residue was dissolved in DCM (15 ml), washed with brine (10 ml), dried with MgSO4, and concentrated in vacuo. The product was purified by chromatography on silica gel (EtOAc:petroleum ether 1:1).
2. Biological Assays
2a. Cell Lines
The normal-like epithelial MCF-10A cell line was purchased from American Type Culture Collection (Manassas, VA, USA) (CRL-10781) and was used as a representative for normal breast epithelial cells. The cell line retains many normal traits, including lack of tumorigenicity in nude mice, anchorage-dependent growth, and dependence on growth factors and hormones for proliferation and survival.19,20 The MCF-10A cells were cultured in RPMI 1640 medium supplemented with 10 % heat-inactivated fetal calf serum (FCS), non-essential amino acids (1 mM), insulin (10 µg/ml), epidermal growth factor (20 ng/ml), cholera toxin (50 ng/ml), hydrocortisol (250 ng/ml), penicillin (100 U/ml), and streptomycin (100 µg/ml).
The JIMT-1 human breast carcinoma cell line (ACC589) was purchased from the German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany). It carries an amplified HER-2 oncogene and is insensitive to HER-2 inhibiting drugs and belongs to the HER2 sub-type of breast cancer.21,22 The JIMT-1 cells were routinely cultured in DMEM/Ham’s F-12 medium supplemented with 10 % FCS, non-essential amino acids (1 mM), insulin (10 µg/ml), penicillin (100 U/ml) and streptomycin (100 µg/ml).
Both cell lines were kept at 37 °C in a humidified incubator with 5 % CO2 in air. For the experiments, cells were seeded at the following densities: MCF-10A: 104 cells/cm2, and JIMT-1: 1.5×104 cells/cm2, in tissue culture vessels of the appropriate size to obtain the desired cell number for the different assays. The volume of medium used was 0.2–0.3 ml per cm2. The cells were allowed to attach for 24 hours before the addition of the compounds.23
2b. Compound Solutions
The sample compounds were dissolved in DMSO to 100 mM stock solutions that were kept at -20 °C. Working solutions were diluted in PBS and all had a DMSO concentration of 0.2 %. In the assay, the cells were exposed to PBS with 0.02 % DMSO, or with the respective compounds at 0.1, 0.25, 0.5, 1, 2.5, 5, 10, and 20 µM concentrations.
2c. Dose Response Assay
The dose response to treatment with the compounds was evaluated using an MTT assay, which is based on reduction of MTT in the mitochondria of live cells. The amount of formazan produced is proportional to the number of living cells.22,23
For the assay, cells were trypsinized and counted in a hemocytometer. Aliquots of 180 µl cell suspension containing 3000 cells (MCF-10A) or 5000 cells (JIMT-1) were seeded in 96-well plates. Compounds were added 24 h later to the final concentrations described above. At 72 h of drug treatment, 20 µl of MTT solution (5 mg/ml in PBS) was added to each well and the 96-well plates were returned to the CO2 incubator for 1 h. The medium was then removed and the blue formazan crystals were dissolved by adding of 100 µl of 100 % DMSO per well. The plates were swirled gently at room temperature for 10 minutes to dissolve the crystals in the cells. Absorbance was monitored at 540 nm in a Multiskan™ FC Microplate Photometer (Thermo Fisher Scientific, Lund, Sweden) using the software SkanIt 3.1. For each compound, three dose response experiments were performed with six replicates for each one of them. GraphPad Prism version 6.01 for Windows (GraphPad Software, La Jolla, CA, USA), was used for drawing dose response curves and calculating the IC50 values, i.e. the dose giving 50 % reduction in cell number.23
Materials and Methods
3. The Preparation of the Assayed Compounds 3a - 3u.
3a. (E)-3-(Benzyliden)Damsin (3a)
3a was obtained as a colourless oil in 72 % yield after chromatographic purification of the product obtained after the Claisen-Schmidt condensation of 4a with 1 under acidic conditions as described in Experimental Procedure (EP) section 1a. (α)D20 +8.1 (c 1.00, CH2Cl2); IR spectrum (film, γ, cm-1): 3366, 3059, 2926, 2870, 1755, 1714, 1623, 1573, 1449, 1271, 1232, 1186, 1158, 1111, 983, 768, 733, 714, 693, 646; TOFMS: [M+H+], found 337.1832, C22H25O3 requires 337.1804. See Table 2 and Table 3 for 1H and 13C NMR shifts.
3b. (E)-3-(4-Methylbenzyliden)Damsin (3b)
3b was obtained as a colourless oil in 70 % yield after chromatographic purification of the product obtained after the Claisen-Schmidt condensation of 4b with 1 under acidic conditions as described in EP section 1a. (α)D20 +29 (c 1.00, CH2Cl2); IR spectrum (film, γ, cm-1): 2921, 2862, 1752, 1712, 1625, 1603, 1271, 1253, 1179, 1159, 1117, 119, 814, 743, 521; TOFMS: [M+H+], found 351.1922, C23H27O3 requires 351.1915. See Table 2 and Table 3 for 1H and 13C NMR shifts.
3c. (E)-3-(3-Methylbenzyliden)Damsin (3c)
3c was obtained as a colourless oil in 90 % yield after chromatographic purification of the product obtained after the Claisen-Schmidt condensation of 4c with 1 under acidic conditions as described in EP section 1a. (α)D20 +3.0 (c 1.00, CH2Cl2); IR spectrum (film, γ, cm-1): 2922, 1758, 1714, 1625, 1271, 1157, 1118, 993, 693; TOFMS: [M+H+], found 351.1933, C23H27O3 requires 351.1915. See Table 2 and Table 3 for 1H and 13C NMR shifts.
3d. (E)-3-(2-Methylbenzyliden)Damsin (3d)
3d was obtained as a colourless oil in 90 % yield after chromatographic purification of the product obtained after the Claisen-Schmidt condensation of 4d with 1 under acidic conditions as described in EP section 1a. (α)D20 +62.6 (c 1.00, CH2Cl2); IR spectrum (film, γ, cm-1): 2922, 2862, 1757, 1714, 1623, 1597, 1270, 1251, 1161, 1119, 982, 949, 759, 735; TOFMS: [M+H+], found 351.1934, C23H27O3 requires 351.1915. See Table 2 and Table 3 for 1H and 13C NMR shifts.
3e. (E)-3-(2,4-Dimethylbenzyliden)Damsin (3e)
3e was obtained as a colourless oil in 46 % yield after chromatographic purification of the product obtained after the Claisen-Schmidt condensation of 4e with 1 under acidic conditions as described in EP section 1a. (α)D20 +12.2 (c 1.00, CH2Cl2);IR spectrum (film, γ, cm-1); 2922, 2857, 1758, 1713, 1625, 1606, 1448, 1384, 1336, 1271, 1241, 1190, 1162, 1119, 1068, 984, 816. TOFMS: [M+H+], found 365.2109, C24H29O3 requires 365.2117. See Table 2 and Table 3 for 1H and 13C NMR shifts.
3f. (E)-3-((4-Trifluoromethylbenzyliden)Damsin (3f)
3f was obtained as a colourless oil in 88 % yield after chromatographic purification of the product obtained after the Claisen-Schmidt condensation of 4-(trifluoromethyl)benzaldehyde with 1 under acidic conditions as described in EP section 1a. (α)D20 +0.9 (c 1.00, CH2Cl2);IR spectrum (film, γ, cm-1): 2925, 2863, 1758, 1717, 1630, 1322, 1163, 1116, 1068, 1014, 984; TOFMS: [M+H+], found 405.1657, C23H24F3O3 requires 405.1633. See Table 2 and Table 3 for 1H and 13C NMR shifts.
3g. (E)-3-((3-Trifluoromethylbenzyliden)Damsin (3g)
3g was obtained as a colourless oil in 56 % yield after chromatographic purification of the product obtained after the Claisen-Schmidt condensation of 4g with 1 under basic conditions as described in EP section 1b. (α)D20 -6.7 (c 1.00, CH2Cl2);IR spectrum (film, γ, cm-1): 2924, 2864, 1758, 1716, 1630, 1327, 1271, 1160, 1073, 1119, 1000, 986, 808, 696, 736; TOFMS: [M+H+], found 405.1655 C23H24F3O3 requires 405.1633. See Table 2 and Table 3for 1H and 13C NMR shifts.
3h. (E)-3-((2-Trifluoromethylbenzyliden)Damsin (3h)
3h was obtained as a colourless oil in 88 % yield after chromatographic purification of the product obtained after the Claisen-Schmidt condensation of 4h with 1 under acidic conditions as described in EP section 1a. (α)D20 +36.8 (c 1.00, CH2Cl2);IR spectrum (film, γ, cm-1): 2925, 1758, 1719, 1631, 1486, 1452, 1387, 1335, 1313, 1286, 1271, 1252, 1154, 1116, 1059, 1034, 985, 814, 799, 769, 735, 666; TOFMS: (M+H+), found 405.1634 C23H24F3O3 requires 405.1633. See Table 2 and Table 3 for 1H and 13C NMR shifts.
3i. (E)-3-(4-Methoxybenzyliden)Damsin (3i)
3i was obtained as a colourless oil in 87 % yield after chromatographic purification of the product obtained after the Claisen-Schmidt condensation of 4i with 1 under acidic conditions as described in EP section 1a. (α)D20 +54.4 (c 1.00, CH2Cl2); IR spectrum (film, γ, cm-1): 2942, 2902, 2869, 2846, 1752, 1709, 1623, 1592, 1510, 1272, 1254, 1179, 1148, 1122, 1027, 979, 940, 835, 814; TOFMS: [M+H+], found 367.1891 C23H27O4 requires 367.1865. See Table 2 and Table 3 for 1H and 13C NMR shifts.
3j. (E)-3-(3-Methoxybenzyliden)Damsin (3j)
3j was obtained as a colourless oil in 73 % yield after chromatographic purification of the product obtained after the Claisen-Schmidt condensation of 4j with 1 under acidic conditions as described in EP section 1a. (α)D20+2.4 (c 1.00, CH2Cl2); IR spectrum (film, γ, cm-1): 2958, 2862, 1752, 1709, 1623, 1592, 1510, 1272, 1254, 1179, 1148, 1122, 1027, 979, 940, 835; TOFMS: [M+H+], found 367.1856 C23H27O4 requires 367.1865. See Table 2 and Table 3 for 1H and 13C NMR shifts.
3k. (E)-3-(2-Methoxybenzyliden)Damsin (3k)
3k was obtained as a colourless oil as the single product obtained after the Claisen-Schmidt condensation of 4k with 1 under acidic conditions as described in EP section 1a. (α)D20 +58.2 (c 1.00, CH2Cl2);IR spectrum ( film, γ, cm-1): 2928, 2859, 1757, 1712, 1619, 1596, 1486, 1463, 1437, 1270, 1247, 1188, 1161, 1119, 984, 814, 754, 743; TOFMS: [M+H+], found 367.1849 C23H27O4 requires 367.1865. See Table 2 and Table 3 for 1H and 13C NMR shifts.
3l (E)-3-(4-Hydroxybenzyliden)Damsin (3l)
4l was protected to 4x according to EP section 1c, 4x was condensed with 1 under basic conditions as described in EP section 1b to form 3x. 3x was subsequently deprotected to 3l according to EP section 1d, 3l was obtained as a colourless oil in 23 % overall yield (from 1) after chromatographic purification of the crude product. (α)D20: +33.0 (c 1.00, CH2Cl2); IR spectrum; 3319, 2923, 2853, 1755, 1737, 1706, 1595, 1578, 1511, 1469, 1443, 1272, 1254, 1168, 1157, 1120,1104, 984, 833, 816; TOFMS: [M+H+], found 353.1722 C22H25O4 requires 353.1708. See Table 2 and Table 3 for 1H and 13C NMR shifts.
3m (E)-3-(3-Hydroxybenzyliden)Damsin (3m)
4m was protected to 4y according to EP section 1c, 4y was condensed with 1 under basic conditions as described in EP section 1b to form 3y. 3y was subsequently deprotected to 3m according to EP section 1d, 3l was obtained as a colourless oil in 43 % overall yield (from 1) after chromatographic purification of the crude product. (α)D20 -11.7 (c 1.00, CH2Cl2); IR spectrum (film, γ, cm-1); 3353, 2927, 2864, 1735, 1712, 1621, 1591, 1578, 1490, 1471, 1449, 1272, 1228, 1158, 1112, 1119, 992, 785, 687; TOFMS: [M+H+], found 353.1733 C22H25O4 requires 353.1708. See Table 2 and Table 3 for 1H and 13C NMR shifts.
3n (E)-3-(2-Hydroxybenzyliden)Damsin (3n)
4n was protected to 4z according to EP section 1c, 4z was condensed with 1 under basic conditions as described in EP section 1b to form 3z. 3z was subsequently deprotected to 3n according to EP section 1d, 3n was obtained as a colourless oil in 27 % overall yield (from 1) after chromatographic purification of the crude product. (α)D20 +29.5 (c 1.00, CH2Cl2); IR spectrum (film, γ, cm-1): 3353, 2927, 2864, 1735, 1712, 1621, 1591, 1578, 1490, 1471, 1449, 1272, 1228, 1158, 1112, 1119, 992, 785, 687; TOFMS: [M+H+], found 353.1704 C22H25O4 requires 353.1708. See Table 2 and Table 3 for 1H and 13C NMR shifts.
3o. (E)-3-Benzyldamsin (3o)
The procedure for preparing 3o is shown in Figure 2. The Michael acceptor functionality of 1 was protected by adding 1 in EtOH (6 ml) to a solution of PhSNa (3.7 eq., 7.4499 mmol) in EtOH (10 ml). The reaction mixture was stirred under N2 atmosphere at room temperature for 3h until completion, quenched with concentrated AcOH (2.5 ml) and H2O (4 ml) at 0 °C. After stirring for an additional 5 minutes, the aqueous phase was extracted with DCM (3 x 10 ml) and the organic layers were dried with Na2SO4, concentrated in vacuo and purified by chromatography on silica gel (EtOAc:petroleum ether 1:1) to yield the protected product 5. This was condensed with benzaldehyde as described above, to give 6. A vial with Pd/C (100 mg) was purged with vacuum/H2 for two cycles to remove air from the reaction tube, whereafter EtOH (2 ml) and a solution of the protected 6 (100 mg, 0.2790 mmol) in DCM (1 ml) and EtOH (1 ml) was added then the mixture of reaction was hydrogenated at ambient pressure and temperature (20 °C) for 29 h. The progress of reaction was followed with TLC. The reaction mixture was filtered in vacuo through a short path of Celite and washed with ethyl acetate (3 ml). The organic layers were concentrated in vacuo to obtain the protected compound 7. For the deprotection, NaIO4 (2.1 eq., 0.0515 mmol) was added to a stirred solution of compound 7 (1 eq., 0.0468 mmol) in MeOH:H2O (1:9, 3 ml) and the reaction mixture was stirred at room temperature for 33 h. When completed it was diluted with DCM (20 ml) and the organic layer was washed with 15 ml of water, whereafter the organic layer were dried with Na2SO4 and concentrated in vacuo to give the elimination product. This mixture of reaction was warmed in PhMe (4 ml) at 120 °C in order to complete the elimination of PhSOH. The crude extract was purified by silica gel chromatography with EtOAc:n-heptane 1:1, to yield 3o with the overall yield 18 % (starting from 1). (α)D20 +36.1 (c 1.00, CH2Cl2);IR spectrum (film, γ, cm-1): 2924, 2870, 1758, 1739, 1625, 1451, 1385, 1338, 1271, 1160, 1118, 1046, 1025, 1002, 981, 949, 815, 753, 699 TOFMS [M+H+], found 339.1960 C22H27O3 requires 339.1960. See Table 2 and Table 3 for 1H and 13C NMR shifts.
3p. (E)-3-(Cyclohexylmethylene)Damsin (3p)
3p was obtained as a colourless oil in 42 % yield after chromatographic purification of the product obtained after the Claisen-Schmidt condensation of 4p with 1 under acidic conditions as described in EP section 1a. (α)D20 -33.6 (c 1.00, CH2Cl2);IR spectrum (film, γ, cm-1): 2923, 2851, 1757, 1720, 1649, 1448, 1384, 1346, 1270, 1250, 1190, 1156, 1101, 993, 953, 802. TOFMS [M+H+], found 343. 2261, C22H31O3 requires 343. 2273. See Table 2 and Table 3 for 1H and 13C NMR shifts.
3q. (Z)-3-(Cyclohexylmethylene)Damsin (3q)
3q was obtained as a colourless oil in 20 % yield after chromatographic purification of the product obtained after the Claisen-Schmidt condensation of 4q with 1 under acidic conditions as described in EP section 1a. (α)D20 +34.4 (c 1.00, CH2Cl2);IR spectrum (film, γ, cm-1): 2924, 2870, 1738, 1660, 1449, 1384, 1341, 1271, 1162, 1119, 1002, 977, 949. TOFMS [M+H+], found 343. 2279 C22H31O3 requires 343. 2273. See Table 2 and Table 3 for 1H and 13C NMR shifts.
3r. (E)-3-Propylidenedamsin (3r)
3r was obtained as a colourless oil in 20 % yield after chromatographic purification of the product obtained after the Claisen-Schmidt condensation of 4r with 1 under acidic conditions as described in EP section 1a. (α)D20 -53.7 (c 1.00, CH2Cl2); IR spectrum (film, γ, cm-1): 2927, 1757, 1649, 1271, 979; TOFMS: [M+H+], found 289.1786, C18H25O3 requires 289.1759. See Table 2 and Table 3 for 1H and 13C NMR shifts.
3s. (E)-3-ButylideneDamsin (3s)
3s was obtained as a colourless oil in 42 % yield after chromatographic purification of the product obtained after the Claisen-Schmidt condensation of 4s with 1 under basic conditions as described in EP section 1b. (α)D20 -44.0 (c 1.00, CH2Cl2); IR spectrum (film, γ, cm-1): 2929, 1758, 1719, 1649, 1269, 1111, 954, 814, 731; TOFMS: [M+H+], found 303.1864, C19H27O3 requires302.1882. See Table 2 and Table 3 for 1H and 13C NMR shifts.
3t. (E)-3-(3-Methylbutylidene)Damsin (3t)
3t was obtained as a colourless oil in 42 % yield after chromatographic purification of the product obtained after the Claisen-Schmidt condensation of 4t with 1 under basic conditions as described in EP section 1b. (α)D20 -63.0 (c 1.00, CH2Cl2); IR spectrum (film, γ, cm-1): 2955, 1757, 1649, 1271, 967; TOFMS: [M+H+], found 317.2100, C20H29O3 requires317.2072. See Table 2 and Table 3 for 1H and 13C NMR shifts.
3u. (E)-3-(Pent-4-en-1-ylidene)Damsin (3u)
3u was obtained as a colourless oil in 36 % yield after chromatographic purification of the product obtained after the Claisen-Schmidt condensation of 4u with 1 under basic conditions as described in EP section 1b. (α)D20 -50.6 (c 1.00, CH2Cl2); IR spectrum (film, γ, cm-1): 2921, 1757, 1719, 1850, 1271, 1112, 994; TOFMS: [M+H+], found 315.1926 C20H27O3 requires315.1915. See Table 2 and Table 3 for 1H and 13C NMR shifts.
Results
Damsin (1) and coronopilin (2) were isolated from the plant Ambrosia arborescens as previously described.14 Since 1 can be obtained in good amounts from the plant, we investigated the possibility to modify the structure of 1 while retaining the α-methylene-γ-lactone moiety intact. The latter is important as procedures that require that this functionality is protected/deprotected would waist too much starting material. The Claisen-Schmidt condensation of 1 with aromatic aldehydes, under either acidic or basic conditions, was found to work reasonably well and without affecting the α-methylene-γ-lactone moiety of 1. The condensation between 1 and benzaldehyde yielded (E)-3-benzylidendamsin (3a), and when the cytotoxicity of 3a towards the two cell lines was compared to that of 1, 3a was found to be more potent in JIMT-1 cells than in MCF-10A cells and thereby more selective for cancer cells than normal cells (see Table 1). Apparently an (E)-3-benzyliden substituent has a favourable effect, either on the reactivity of the α-methylene-γ-lactone moiety, or on the association of 3a with critical cellular components such as proteins. To investigate this further, we decided to expand the investigation and study 3-benzyliden derivatives formed by the Claisen-Schmidt condensation of 1 with the aromatic aldehydes 4a – 4n. The 13 derivatives 3b – 3n (see Figure 1) substituted with methyl , triflouro-, methoxy- and hydroxyl groups were prepared, and assayed for cytotoxicity (see Table 1). In general, the condensations proceeded well, and often, but not always, best under acidic conditions (the yields are given in Materials and Methods). However, it was not possible to use the hydoxybenzaldehydes 4l – 4n directly (to produce 3l – 3n), instead the hydroxyl groups had to be protected as MOMO substituents to form 4x – 4z (see Figure 2) prior to the condensation with 1, to form compounds 3x – 3z (see Figure 2). These were subsequently deprotected as described in the Experimental section, to yield 3l – 3n. The condensations with aromatic aldehydes were highly stereoselective, only the E-alkenes were obtained. To investigate the influence of the double bond resulting from the condensation, the reduced derivative 3o was prepared. As direct hydrogenation of 3a also reduced the C-11/C-13 double bond in the α-methylene-γ-lactone moiety, it was necessary to first protect the C-11/C-13 double bond of 1 as the 13-phenylthio ether to give compound 5 (see Figure 2). 5 was then condensed with benzaldehyde to give 6 which subsequently was reduced to 7 and deprotected to obtain the desired product 3o (see Figure 2 and Materials and Methods). In order to investigate if the 3-substituent needs to be aromatic, the condensation was also carried out with cyclohexanecarbaldehyde. In this aldol condensation both the E (3p) and the Z (3q) isomers were obtained, which gave us an opportunity to investigate the influence of this double bond’s configuration on the activity. Finally, aldol condensations were also performed with a few alkyl- and alkenyl aldehydes, produurecing the derivatives 3r – 3u for comparison.
Table 1. The cytotoxicity of compounds 1, 2, and 3a – 3u (see Figure 1), given as IC50 values (mM) calculated from experiments with six concentrations up to 20 mM. Standard deviations were obtained from 1three dose-response curves or 2four dose-response curves. na, not applicable.| Compound | MCF-10A (µM) | JIMT-1 (µM) | Ratio MCF-10A:JIMT-1 |
| 1 | 8.1 ± 0.42 | 3.3 ± 0.62 | 2.5 |
| 2 | 15.3 ± 0.92 | 5.6 ± 0.82 | 2.7 |
| 3a | 8.2 ± 1.62 | 1.7 ± 0.41 | 4.8 |
| 3b | 3.7 ± 0.42 | 2.1 ± 0.32 | 1.8 |
| 3c | 12.6 ± 1.62 | 4.8 ± 0.32 | 2.6 |
| 3d | 11.1 ± 1.82 | 4.7 ± 0.11 | 2.4 |
| 3e | 5.2 ± 1.52 | 3.5 ± 0.72 | 1.5 |
| 3f | 3.1 ± 0.32 | 1.8 ± 0.22 | 1.7 |
| 3g | 11.9 ± 0.41 | 4.4 ± 0.71 | 2.7 |
| 3h | 13.0 ± 0.81 | 8.1 ± 0.61 | 1.6 |
| 3i | 7.9 ± 1.22 | 1.6 ± 0.12 | 4.9 |
| 3j | > 201 | 9.0 ± 1.01 | na |
| 3k | > 201 | 7.1 ± 0.21 | na |
| 3l | 13.6 ± 0.62 | 2.9 ± 0.21 | 4.7 |
| 3m | 10.6 ± 1.32 | 2.4 ± 0.11 | 4.4 |
| 3n | 6.7 ± 0.92 | 2.1 ± 0.21 | 3.2 |
| 3o | 7.1 ± 0.62 | 2.0 ± 0.62 | 3.6 |
| 3p | 11.7 ± 1.92 | 8.1 ± 3.12 | 1.4 |
| 3q | > 202 | 12.3 ± 1.32 | na |
| 3r | 5.5 ± 1.11 | 1.4 ± 0.11 | 3.9 |
| 3s | 12.6 ± 0.81 | 3.7 ± 0.11 | 3.4 |
| 3t | 20.3 ± 0.31 | 8.1 ± 0.11 | 2.5 |
| 3u | 17.5 ± 5.11 | 1.7 ± 0.01 | 10.3 |
Figure 2. The hydroxybenzaldehydes 4l – 4n were protected using MOMBr and EtN(iPr)2, and the condensations of the protected hydroxybenzaldehydes 4x - 4z with 1 were carried out under basic conditions (3.2.1b). The reduced derivative 3o was prepared by protection of C-13 of 1 as a phenyl thiol ether (5), the condensation of 5 with benzaldehyde under acidic conditions (3.2.1a) to give 6, which was hydrogenated to 7 and deprotected to yield 3o.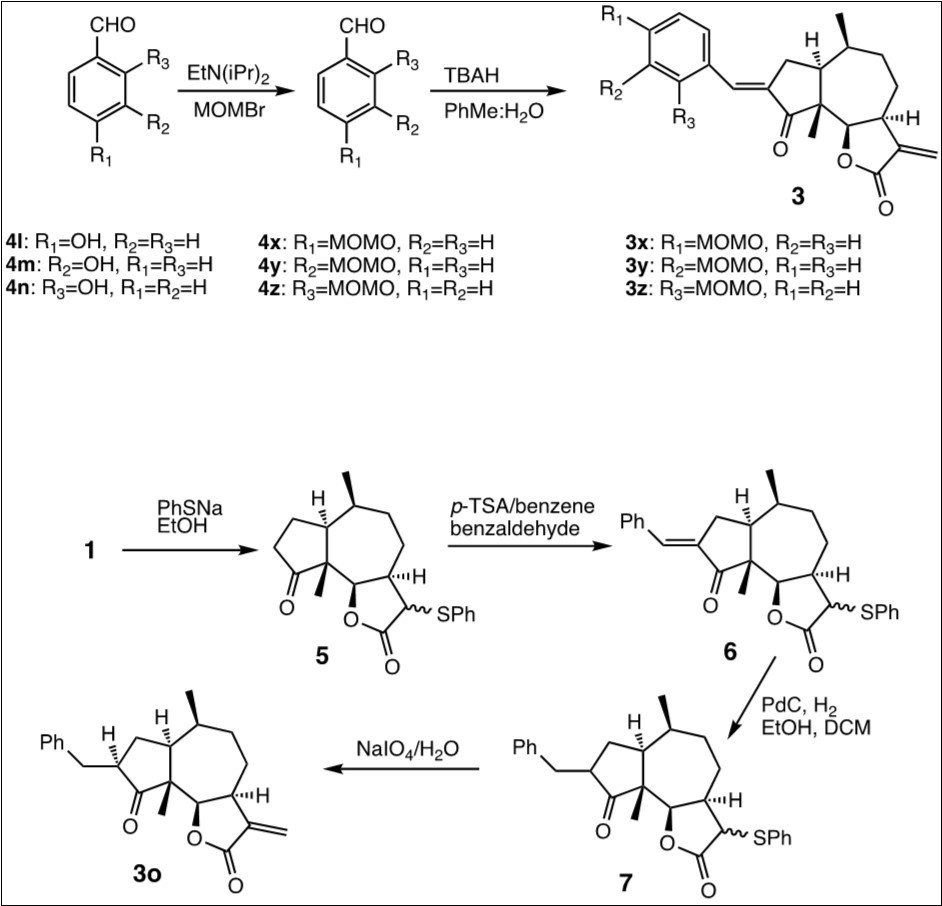
Discussion
The structures of all compounds prepared in this investigation was carefully determined by 1- and 2D NMR experiments (including COSY, NOESY, HMQC and HMBC experiments), in combination with the IR and HRMS data reported in the Experimental section. The configuration of the C-3/C-1' double bond was determined by NOESY NMR experiments. For the E isomers correlations were observed between 2-H2 and 2''-H/6''-H (for 3a - 3n) or 1''-H2 (for 3p - 3u), and between 1'-H and 2-H2 for the Z-isomer 3q. The absolute configuration of C-3 in derivative 3o was suggested by the observed NOESY correlation between 1-H and 3-H, as well as the lack of NOESY correlations between both 14-H3 and 15-H3, and 3-H. This configuration is also supported by the 1H-1H coupling constants observed, 8.4 and 12.1 Hz between 3-H and 2-H2 as well as 3.9 and 8.8 Hz between 3-H and 1'-H2.
The chemical shifts for all proton and carbon signals observed in CDCl3 are presented in Table 2 and Table 3. In all cases, the NMR couplings observed between protons are those that we expected from our previous experience from this type of compounds, and are for reasons of space not reported here. The comparison of the proton and carbon shifts in Table 2 and Table 3 gives some interesting information. Firstly, in all the prepared and assayed compounds except for 3o, a second Michael acceptor functionality (C-1'/C-3/C-4/4-O) is created by the condensation, and one may ask if this also is involved in the chemical reactivity of the compounds. The NMR chemical shifts, as well as the reactivity of the compounds, are strongly influenced by the electronic properties of the chemical bonds close to a specific nucleus or involved in a reactive functionality. Michael acceptors are more reactive if they are perfectly conjugated and situated in a plane, which is reflected in the chemical shifts of the atoms involved. If this new Michael acceptor functionality is well conjugated and thereby reactive, one would expect the carbon shift of the keto functionality (C-4) to be below 200, not close to 210. The exceptions are 3o which has no double bond for conjugation (carbon shift of C-4 218.6), and the Z-isomer 3q in which this Michael acceptor functionality for steric reasons can not be in a plane. The conclusion is therefore that only a weak conjugation is present, and that the reactivity of the C-1'/C-3/C-4/4-O Michael acceptor is small. Secondly, assuming that the α-methylene-β-lactone moiety is critical for the cytotoxicity of all compounds assayed, it is interesting to compare the chemical shifts of 6-H, 7-H, 13-H2, C-6, C-7, C-11, C-12 and C-13 of the assayed compounds, in search for any indication that the substituent at C-3 affect the electronic properties of this lactone ring. 6-H are all between 4.6 and 4.7 ppm, except in 3o and 3q in which the shift is slightly lower (due to the lack of conjugation discussed above). 7-H are all between 3.2 and 3.3 ppm, 13-Ha are between 5.5 and 5.6 ppm, while 13-Hb are between 6.2 and 6.3 ppm. All C-6 values are close to 82 ppm, C-7 between 44.5 and 45.0 ppm, C-11 between 139.6 and 140.2 ppm, C-12 between 170.2 and 171.1 ppm, and C-13 between 120.8 and 121.9. So, the electronic properties of the α-methylene-β-lactone moiety remains essentially intact, and independent of the C-3 substituent. The conclusion is therefore that the differences in cytotoxic activities shown in Table 1 and discussed below, depend on the molecular interaction that the C-3 substituent can provide with a protein at the site where a Michael addition can take place.
Table 2. 1H NMR chemical shifts () for the assayed compounds 3a – 3u, determined at 400 MHz in CDCl3.| 1-H | 2-H2 | 6-H | 7-H | 8-H2 | 9-H2 | 10-H | 13-H2 | 14-H3 | 15-H3 | 1’-H | 2’’-H | 3’’-H | 4’’-H | 5’’-H | 6’’-H | |
| 3a | 2.07 | 2.87/2.97 | 4.65 | 3.28 | 1.80/2.06 | 1.74/1.89 | 2.28 | 5.57/6.28 | 1.17 | 1.16 | 7.44 | 7.41 | 7.54 | 7.37 | 7.54 | 7.41 |
| 3ba | 2.06 | 2.84/2.94 | 4.63 | 3.38 | 1.80/2.07 | 1.74/1.88 | 2.27 | 5.56/6.27 | 1.17 | 1.14 | 7.4 | 7.43 | 7.21 | - | 7.21 | 7.43 |
| 3cb | 2.05 | 2.84/2.94 | 4.63 | 3.28 | 1.80/2.06 | 1.73/1.87 | 2.28 | 5.56/6.26 | 1.17 | 1.14 | 7.38 | 7.32 | - | 7.17 | 7.3 | 7.32 |
| 3dc | 2.03 | 2.74/2.94 | 4.64 | 3.28 | 1.81/2.06 | 1.70/1.86 | 2.24 | 5.57/6.28 | 1.16 | 1.18 | 7.65 | - | 7.23 | 7.26 | 7.24 | 7.46 |
| 3ed | 2.03 | 2.75/2.94 | 4.65 | 3.28 | 1.82/2.09 | 1.70/1.86 | 2.24 | 5.57/6.29 | 1.19 | 1.16 | 7.66 | - | 7.07 | - | 7.07 | 7.38 |
| 3f | 2.1 | 2.86/2.99 | 4.66 | 3.31 | 1.84/2.07 | 1.74/1.91 | 2.3 | 5.58/6.29 | 1.19 | 1.17 | 7.43 | 7.65 | 7.66 | - | 7.66 | 7.65 |
| 3g | 2.1 | 2.85/2.98 | 4.65 | 3.3 | 1.84/2.08 | 1.75/1.91 | 2.31 | 5.58/6.29 | 1.19 | 1.17 | 7.43 | 7.76 | - | 7.62 | 7.55 | 7.71 |
| 3h | 2.05 | 2.65/2.90 | 4.64 | 3.29 | 1.81/2.03 | 1.72/1.85 | 2.22 | 5.56/6.27 | 1.14 | 1.18 | 7.71 | - | 7.58 | 7.58 | 7.45 | 7.71 |
| 3ie | 2.06 | 2.82/2.92 | 4.63 | 3.27 | 1.79/2.04 | 1.74/1.86 | 2.27 | 5.56/6.26 | 1.16 | 1.13 | 7.37 | 7.5 | 9.93 | - | 6.93 | 7.5 |
| 3jf | 2.06 | 2.86/2.95 | 4.64 | 3.28 | 1.80/2.03 | 1.72/1.86 | 2.27 | 5.57/6.27 | 1.16 | 1.15 | 7.38 | 7.05 | - | 6.92 | 7.33 | 7.14 |
| 3kg | 2.01 | 2.73/2.91 | 4.59 | 3.25 | 1.77/2.04 | 1.72/1.84 | 2.22 | 5.55/6.24 | 1.14 | 1.14 | 7.82 | - | 6.89 | 7.32 | 6.96 | 7.47 |
| 3l | 2.07 | 2.81/2.91 | 4.67 | 3.3 | 1.81/2.04 | 1.72/1.90 | 2.27 | 5.59/6.29 | 1.16 | 1.14 | 7.36 | 7.43 | 6.93 | - | 6.93 | 7.43 |
| 3m | 2.01 | 2.80/2.90 | 4.71 | 3.34 | 1.84/2.04 | 1.74/1.90 | 2.24 | 5.59/6.29 | 1.14 | 1.12 | 7.48 | 7.08 | - | 6.91 | 7.25 | 7.25 |
| 3n | 2.04 | 2.79/2.94 | 4.64 | 3.29 | 1.82/2.06 | 1.73/1.87 | 2.24 | 5.58/6.29 | 1.16 | 1.17 | 7.92 | - | 6.94 | 7.22 | 6.9 | 7.45 |
| 3oh | 1.99 | 1.71/1.87 | 4.5 | 3.31 | 1.79/2.02 | 1.69/1.81 | 2.16 | 5.53/6.29 | 1 | 0.91 | 2.66/3.19 | 7.17 | 7.28 | 7.21 | 7.28 | 7.17 |
| 3pi | 2 | 2.51/2.59 | 4.59 | 3.26 | 1.78/2.03 | 1.71/1.84 | 2.23 | 5.54/6.26 | 1.1 | 1.08 | 6.47 | 1.23/1.66 | 1.26/1.75 | 1.77 br | 1.26/1.75 | 1.23/1.66 |
| 3qj | 2.01 | 1.82/2.55 | 4.47 | 3.28 | 1.82/2.03 | 1.72/1.80 | 2.21 | 5.52/6.26 | 1.05 | 1.03 | 5.43 | 1.59/2.08 | 1.65/1.97 | 1.71 br | 1.65/1.97 | 1.59/2.08 |
| 3rk | 2 | 2.50/2.58 | 4.6 | 3.26 | 1.79/2.02 | 1.71/1.86 | 2.23 | 5.55/6.26 | 1.11 | 1.09 | 6.61 | 2.16 | 1.06 | - | - | - |
| 3sl | 1.98 | 2.47/2.53 | 4.56 | 3.24 | 1.76/2.00 | 1.68/1.79 | 2.19 | 5.52/6.20 | 1.06 | 1.04 | 6.57 | 2.09 | 1.44 | 0.88 | - | - |
| 3tm | 1.96 | 2.50/2.54 | 4.59 | 3.25 | 1.74/1.86 | 1.70/1.84 | 2.19 | 5.53/6.23 | 1.07 | 1.05 | 6.62 | 2.02 | 1.76 | 0.89 | 0.89 | - |
| 3un | 2 | 2.47/2.55 | 4.6 | 3.26 | 1.78/2.02 | 1.73/1.85 | 2.24 | 5.51/6.26 | 1.09 | 1.08 | 6.61 | 2.25 | 2.22 | 5.78 | 5 | - |
| C-1 | C-2 | C-3 | C-4 | C-5 | C-6 | C-7 | C-8 | C-9 | C-10 | C-11 | C-12 | C-13 | C-14 | C-15 | C-1’ | C-1’’ | C-2’’ | C-3’’ | C-4’’ | C-5’’ | C-6’’ | |
| 3a | 43.6 | 31.4 | 133.3 | 207.9 | 54.8 | 81.8 | 44.8 | 26.5 | 34.2 | 34.1 | 140.1 | 170.2 | 121.2 | 15.7 | 14.5 | 133.9 | 129 | 130.6 | 128.8 | 135.4 | 128.8 | 130.6 |
| 3ba | 43.6 | 31.4 | 132.4 | 208.1 | 54.8 | 81.9 | 44.8 | 26.5 | 34.3 | 34.1 | 140.1 | 170.3 | 121.3 | 15.8 | 14.5 | 133.9 | 132.6 | 130.6 | 129.6 | 140 | 129.6 | 130.6 |
| 3cb | 43.6 | 31.4 | 133.2 | 208.1 | 54.8 | 81.9 | 44.8 | 26.5 | 34.2 | 34 | 140.1 | 170.3 | 121.3 | 15.8 | 14.5 | 134 | 135.3 | 131.4 | 138.4 | 130.4 | 128.7 | 127.6 |
| 3dc | 43.9 | 31.3 | 134.1 | 207.9 | 54.9 | 81.9 | 44.8 | 26.4 | 34.1 | 34 | 140.1 | 170.3 | 121.2 | 15.8 | 14.5 | 131.5 | 134.1 | 139 | 130.7 | 129.4 | 125.9 | 128.7 |
| 3ed | 44 | 31.5 | 133.3 | 208 | 55 | 82 | 45 | 26.5 | 34.3 | 34.2 | 140.2 | 170.4 | 121.2 | 15.9 | 14.6 | 131.7 | 131.4 | 139.3 | 131.6 | 139.8 | 126.7 | 128.8 |
| 3fe | 43.6 | 31.4 | 135.8 | 207.6 | 55 | 81.7 | 44.8 | 26.4 | 34.1 | 34 | 139.9 | 170.2 | 121.5 | 15.7 | 14.4 | 132 | 138.9 | 130.6 | 125.7 | 130.8 | 125.7 | 130.6 |
| 3gf | 43.7 | 31.2 | 135.2 | 207.7 | 54.9 | 81.8 | 44.8 | 26.4 | 34.2 | 34 | 140 | 170.3 | 121.4 | 15.7 | 14.5 | 132 | 131.4 | 126.8 | 136.2 | 125.9 | 129.4 | 133.5 |
| 3hg | 43.7 | 31.1 | 137 | 207 | 55 | 81.7 | 44.7 | 26.3 | 34 | 34 | 139.9 | 170.3 | 121.3 | 15.7 | 14.4 | 129.4 | 129.8 | 134.1 | 130.1 | 131.7 | 128.8 | 126.3 |
| 3ih | 43.6 | 31.4 | 130.9 | 208 | 54.7 | 81.9 | 44.9 | 26.6 | 34.3 | 34.1 | 140.2 | 170.3 | 121.2 | 15.8 | 14.6 | 133.7 | 128.1 | 132.4 | 114.3 | 160.7 | 114.3 | 132.4 |
| 3ji | 43.6 | 31.4 | 133.7 | 208 | 54.8 | 81.8 | 44.8 | 26.5 | 34.2 | 34 | 140.1 | 170.3 | 121.3 | 15.7 | 14.5 | 133.8 | 136.7 | 116.1 | 159.7 | 115 | 129.8 | 123.1 |
| 3kj | 43.6 | 31.4 | 133.2 | 207.9 | 54.7 | 81.9 | 44.8 | 25.4 | 34.1 | 34 | 140.1 | 170.4 | 121.1 | 15.8 | 14.5 | 128.5 | 124.3 | 158.8 | 110.8 | 131.1 | 120.2 | 129.6 |
| 3l | 43.8 | 31.4 | 130.3 | 208.7 | 54.8 | 82.3 | 44.8 | 26.5 | 34.3 | 34.1 | 140.1 | 170.9 | 121.7 | 15.8 | 14.6 | 134.7 | 127.5 | 132.8 | 116.1 | 158.2 | 116.1 | 132.8 |
| 3m | 43.6 | 31.5 | 133.3 | 208.8 | 54.9 | 82.4 | 44.5 | 26.3 | 34.1 | 34 | 139.9 | 171.1 | 121.9 | 15.7 | 14.5 | 134.8 | 136.8 | 122 | 156.7 | 117.3 | 129.8 | 118.5 |
| 3n | 43.8 | 31.4 | 132 | 209.1 | 55 | 82.1 | 44.8 | 26.5 | 34.2 | 34.1 | 140.1 | 170.7 | 121.5 | 15.8 | 14.6 | 129.8 | 122.5 | 156.8 | 116.4 | 131.5 | 120 | 129.6 |
| 3o | 44.1 | 30.6 | 50.3 | 218.6 | 55.3 | 81.9 | 44.5 | 25.6 | 33.1 | 34.2 | 139.6 | 170.3 | 120.8 | 14.6 | 14 | 36.3 | 139.4 | 129.2 | 128.5 | 126.4 | 128.5 | 129.2 |
| 3p | 43.2 | 28.6 | 132.8 | 207.6 | 55.4 | 81.9 | 44.9 | 26.5 | 34.2 | 34.2 | 140.2 | 170.4 | 121.1 | 15.8 | 14.4 | 143.1 | 39.1 | 31.7 | 25.6 | 25.1 | 25.6 | 31.7 |
| 3q | 44.1 | 28.3 | 135.4 | 219.9 | 55.3 | 82 | 44.6 | 25.3 | 33.3 | 34.3 | 139.6 | 170.3 | 120.8 | 16.2 | 14.3 | 122.9 | 39.5 | 31.6 | 25.8 | 25.5 | 25.8 | 31.6 |
| 3r | 43.1 | 28.5 | 134.1 | 207.2 | 55.5 | 81.9 | 44.9 | 26.5 | 34.2 | 34.2 | 140.1 | 170.3 | 121.2 | 15.8 | 14.4 | 139.7 | 23.2 | 12.9 | - | - | - | - |
| 3s | 43 | 28.6 | 134.8 | 207 | 55.3 | 81.8 | 44.7 | 26.3 | 34 | 34 | 140 | 170.3 | 121.1 | 15.7 | 14.2 | 138.1 | 31.7 | 21.6 | 13.9 | - | - | - |
| 3t | 43 | 28.8 | 135.3 | 207 | 55.4 | 81.9 | 44.7 | 26.4 | 34.1 | 34.1 | 140.1 | 170.4 | 121.2 | 15.7 | 14.3 | 137.4 | 38.9 | 28.2 | 22.6 | 22.6 | - | - |
| 3u | 43.1 | 28.7 | 135.1 | 206.9 | 55.5 | 81.8 | 44.8 | 26.4 | 34.2 | 34.2 | 140.1 | 170.3 | 121.2 | 15.7 | 14.4 | 137.3 | 29.2 | 34.3 | 137.3 | 115.6 | - | - |
Conclusion
The MCF-10A cell line used in this study is a non-tumorigenic breast epithelial cell line15 while the JIMT-1 cells are breast cancer-derived tumorigenic.16 Table 1 shows the experimental cytotoxicity IC50 values in M obtained with the two cell lines, calculated from the dose response curves. In addition, the ratios between the IC50 MCF-10A and IC50 JIMT-1 were calculated, and are presented in Table 1 as a measure for the selectivity of each compound. As indicated above, (E)-3-benzylidendamsin (3a) possessing a phenylmethylene group in position 3, has a similar cytotoxicity as 1 in MCF-10A cells although it is more potent in JIMT-1 cells, and this simple improvement of potency and selectivity was the starting point for this investigation. The IC50 values show consistently that the JIMT-1 cells are more sensitive to the compounds assayed compared to the MCF-10A cells. It is interesting to compare the toxicity with the population doubling time of the two cell lines. For MCF-10A cells this is approximately 17 h, while for JIMT-1 cells it is 24 h. Consequently, the MCF-10A cells have had a longer exposure time since they have had the possibility to go through more population doublings, suggesting that the toxicity is not related to the rate of cell proliferation but to some other intrinsic property in the cells. One such property could be the intracellular glutathione level. Glutathione has been shown to reduce the toxicity of sesquiterpene lactones17 and MCF-10A cells have been shown to have a slightly higher glutathione level than JIMT-1 cells.18
The cytotoxicity of the reduced derivative 3o is similar to that of 3a, indicating that the double bond created at C-3 by the condensation is unimportant. Replacing the phenyl group of 3a with a cyclohexyl (in 3p) decreases both potency and selectivity, and when comparing 3p with 3q it is obvious that an E configuration of the C-3 double bond is preferable. None of the methylated derivatives 3b – 3e is more potent compared to 3a, although the p-substituted derivative 3b has a similar potency, and they are all less selective. Methyl substituents in the benzene ring are therefore not beneficial. For the trifluoromethyl derivatives 3f – 3h there is a strong tendency that p-substitution is better for the potency than m-substitution, and m is better than o, but all three suffer from lower selectivity. For the methoxylated derivatives 3i – 3k, it is obvious that the p-methoxy derivative 3i not only retains the potency of 3a but also the selectivity. However, the m- and o-methoxy derivatives 3j and 3k are considerably less cytotoxic. Furthermore, the p-hydroxy derivative 3l retains much of the potency and selectivity of 3a, although the m- and o-hydroxyl derivatives 3m and 3n are even more potent than 3l. The differences in the trends observed may be explained by the ability of the hydroxyl group to both give and accept hydrogen bonds, which is not the case for the other substituents used here. Also, the size of a hydroxyl group is smaller than a trifluoromethyl or methoxy group, so there may be a steric component. For the alkenyl derivatives 3r – 3u, the results are quite interesting. The smallest, 3r, is as potent and selective as 3a, indicating that a short alkyl group influences the molecular interaction as much as a phenyl group. An extra methyl group (as in 3s) is less advantageous, while two extra methyl groups (as in 3t) is even worse for both potency and selectivity. Remarkably, by keeping the same number of carbons in the chain but without branching and with a double bond in the end (as in 3u), the potency is the same as that of 3a while the selectivity is high. With a ratio of IC50 MCF-10A cells and IC50 JIMT-1 cells greater than 10, 3u is the most selective derivative prepared in this investigation, and will be a future starting point for the development of this class of compounds.
Acknowledgments
The authors thank the Swedish International Development Agency (SIDA) for the financial support of this study, which is part of the project “Biomolecules of medicinal and industrial interest” developed between the Universidad Mayor de San Andrés (UMSA La Paz – Bolivia) and Lund University (Sweden).