Abstract
With the definition of four gene classes, all differences between tumor cells and normal cells can be explained. Proliferative mutations induce a shortcut, forcing the cell to divide. They allow replication without control, induce somatic pairing defects of chromosomes and genome instability. Intact Tumor Supressors or mutant Switch Functions can inhibit this process. Oncogene mutations optimize the growth of the cells.
Author Contributions
Academic Editor: Xin Qing, Head, Hematopathology Director, Hematology and Flow Cytometry Laboratory, United States.
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2019 Isolde Riede
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Introduction
In about the last 40 years, a time with efforts in molecular biology and genetic engineering a variety of gene classes involved in tumor formation, cell division and cell death was defined. Oncogenes (ONC) were the first class of genes analyzed, defined by Huebner and Todaro in 1969. They postulated from cell culture experiments, that “A viral information is responsible for transforming a normal cell into a tumor cell (the oncogene)” 1. ONC seemed to actively induce cell proliferation. Since then, a large variety of ONC were found and analyzed. ONC mutations are dominant, i.e. one copy of it is sufficient for the transformation of the cell. Human genetics illuminated predispositions for familial tumors. In attempts to find the respective genes, TP53 defects were found as the cause for inherited retinoblastoma. With the intensive research on TP53, the definition for tumor suppressor genes (TUS) found its way into the scientific community in 1983 2. In the basic experiment, cells from tumor origin were fused to normal cells. This resulted in cells that did not proliferate. It was concluded, that a factor of the non tumor cell should inhibit growth potential of the tumor cell 3. The absence of a tumor suppressor function allows cell growth. Based on experiments in cell culture, most general features were defined.
Proliferative Genes (PRO) were defined in 1997 due to experiments in Drosophila 4. Their mutation results in somatic pairing defects of chromosomes, and therefore they influence a general feature of the chromosome structure. Later, ALL-1, a gene causative for human leukemia, was identified as a proliferative gene 5. Mutant proliferative genes allow replication without control. This always induces new cell divisions with no possible growth arrest in a cell phase, where the specific cellular functions are expressed.
Switch genes (SWI) were as well defined from experiments in Drosophila 6. Mutant forms are able to inhibit cell proliferation induced by a proliferative gene. Their inhibition in tumor cells can lead to growth arrest of the tumor (Table 1).
Table 1. Overview of functions involved in tumor formation| wildtype in normal cell | mutant in tumor cell | |
| ONC | cellular metabolism functions | enhance growth of cell |
| TUS1 | decision for cell arrest or apoptosis | no cell arrest, growth of cell |
| PRO | coordination of replication | allow replication without control shortcut of the cell cycle |
| SWI | master genes of function complexes | not mutant in tumor cell, but overexpressed; allow tumor growth |
ONC
Research on ONC was intensified around 1980 7, 8. Molecular identification of several new genes involved in enhancement of cell growth led to a large variety of biochemical functions. Basic experiment for many ONC identifications was the appearance of foci in cell culture (faster growing cells by mutation), some ONC were found in animal systems. More and more genes were identified, and nearly all biochemical areas of the cell metabolism are involved. For example, ras is a small GPTase, which plays a role as a central component controlling a variety of signalling pathways in the cell 9; or fos was identified as transcription factor 10; or erbA emerged as thyroid hormone receptor 11. Until today, no complete picture is visible, on how these genes combined together could contribute to cancer growth. Therefore, in 1988 Graf postulated that leukemia formation is a multistep transformation 12.
In contrast to this multistep process, cytogenetic studies of leukemia cells had revealed single rearrangements of chromosomes as cause of the tumor growth. This led to the hypothesis that single events can cause tumor formation. All-1 mutations participate in acute leukemia in 5% to 10% of children and adults. Like other genes, All-1 was discovered by the association with chromosome rearrangements, involving its location in chromosome band 11q23 13.
However, the research on ONC revealed a large variety of possible involvements in different aspects of the process of tumor formation. During tumor initiation, the microenvironment around the tumor cells is considered to play an important role. In this phase, the tumor cells hide from the immune system, and myc could play an important role in this behavior 14. myc coordinates the expression of many genes involved in cell proliferation and differentiation. It activates as well immune regulatory cytokines and the production of the immune checkpoint protein CD47. Other oncogenes, that modulate myc, have as well been shown to regulate immune checkpoints. myc could prevent the induction of an immune response by complex interactions within the cell and by modulating immune cells. It is postulated, that myc suppression in combination with immunregulatory interventions could be used in the future in tumor therapies.
The microenvironment as well might play a role in tumor initiation and promotion 15. Here, inflammatory cells are important, which enhance cell proliferation of tumor cells with cytokines. Epithelial cells can reprogram to inflammatory cells on their own. In this way they get a less differentiated state and gain a higher proliferative potential. This opens the tissue and allows tumor tissue to grow.
TUS
TUS were defined 1974 by Drosophila genetics 16. Human genetics long ago mapped genetic disposition of retinoblastoma. This lead to the identification of TP53, causing a tumor when absent 17. TP53 thereby revealed as differing from ONC: ONC induce tumors following activation or alteration. Here the absence of both alleles on both chromosomes allows the development of retinoblastoma. An additional proliferative factor was postulated, that induces the tumor. TP53 was identified to be involved in the decision point in case of DNA damage for cell arrest and DNA repair (in case of low damage) or apoptosis (in case of high DNA damage) 18. Further research on human tumor disposition genes, revealed the involvement of the repair system itself in the process of tumor formation 19, 20.
TP53 is known to be the most frequently mutated gene across the majority of human cancer types. Therapy resistance to cytotoxic treatments is one of the consequences 21. In premalignant and malignant tissues, TP53 can be absent in some cells, or over-expressed in others, meaning a heterogeneity of gene expression levels. This is partially caused by variations in gene copy number, obviously caused by genome instability. Today, TP53 tests begin to become a routine in specialized tumor pathology laboratories, with results in consequences for therapy decisions.
PRO
Mutant proliferative genes in Drosophila appear with melanotic tumors in larvae. These mutations hinder chromosome pairing in distinct chromosome regions. They allow replication without control, which shortcuts the cell cycle. They as well can induce genome plasticity.
Cells usually enter a G1 (gap 1) phase after mitosis (Figure 1). This is the working phase for each cell type, and is the time when cell specific biochemical functions are executed. Subsequently, the cells may enter a G0 phase, meaning that the cell reaches its final differentiation, with no remaining ability to divide. G1 or G0 phase is shortcut by a proliferative mutation, which allows replication without restriction. After the S (synthesis of DNA) phase, cells go through a short G2 phase, which cannot be modulated or shaped, but is fixed in time and biochemical action to prepare the cell division (M, mitosis). Proliferative genes allow replication; this leads to the S phase directly, with no restriction point to arrest the tumor cell.
Figure 1. The circle represents the cell cycle (explanation in the main text). The red arrow shows the action of a proliferative mutation, allowing replication. This shortcuts the cell cycle forcing the cell to always new cell divisions. 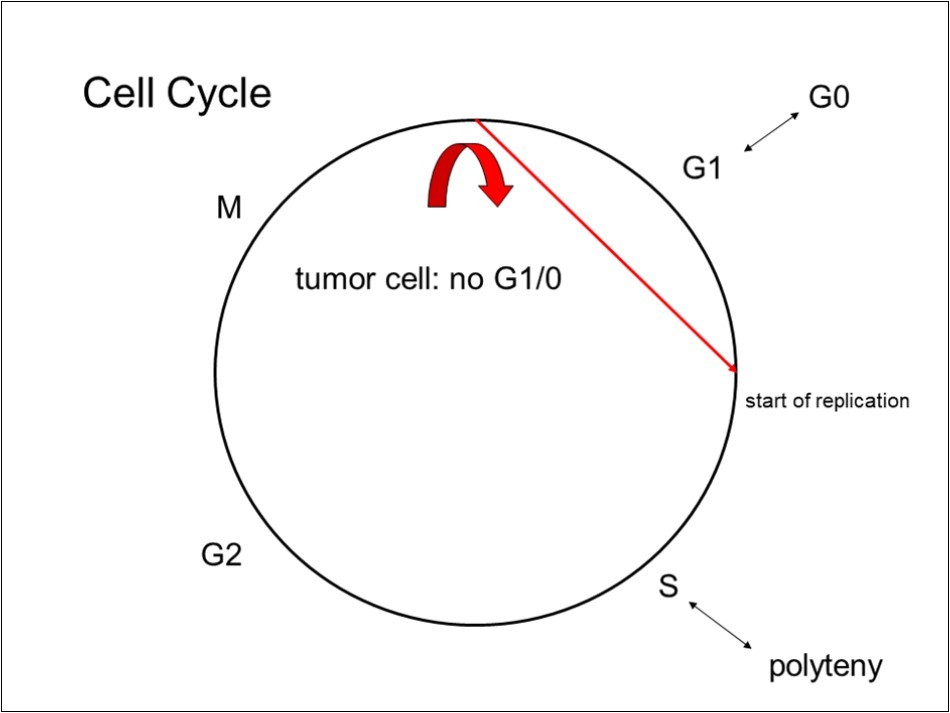
All-1 mutation is a proliferative gene defect; cells allow replication without regulation directly after mitosis 5. A biochemical study revealed, that this aberrant onset of replication includes onset of the repair system, and that apoptosis cannot be induced by high DNA damage.
Cytogenetics and biochemical analyses revealed that one single proliferative mutation
allows replication
and as a result shortcuts the cell cycle
allows tumor growth of cells
allows metastatic behavior of cells
shows somatic pairing defects
shows recombination irregularities
induces genome instability
induces loss of telomers
induces the repair system
induces apoptosis defect
induces intrinsic resistance to chemotherapeutic drugs 22.
Especially the aspect of genome instability causes major problems in tumor therapy, because cells of one tumor tissue are not always clonal but differ from each other. Aneuploidy, or imbalanced chromosome number, is one of the consequences of the instability. This has profound effects on human cells. Physiological changes include altered cell growth, transcriptional changes and further genomic instability 23. These effects add to further loss of differentiation of the tumor cell and a higher proliferative potential.
SWI
SWI were defined by Drosophila genetics 6. Mutant forms of SWI can inhibit the proliferative potential of PRO. They all carry HOX boxes and each regulates the expression level of about 50 genes. In human tumor cells, SWI are over-expressed 24. It is postulated, that a self regulation process activates them to optimize the cell growth. Therefore they could regulate general features in the cell in synthesizing molecules for replication and mitosis. As well in the human system, the down regulation of SWI reduces cell proliferation 25. Their down regulation in patients can reduce tumor growth 22.
Conclusion
Proliferation of tumor cells is due to PRO mutations that allow replication. This first biochemical change induces the change of cellular functions, which then focus on the support of cell divisions. ONC and SWI are involved to support this division process, missing TUS allow the cell divisions.