Abstract
Progressive multifocal leukoencephalopathy (PML) is a rare complication associated, inter alia, with rituximab-based lymphoma treatment. PML diagnosis is made easier with the criteria recently published by the American Academy of Neurology. Unambiguous diagnosis of PML can be achieved by demonstration of the histopathological triad comprising:(1) demyelination, (2) bizarre astrocytes and (3) enlarged oligodendroglial nuclei together with detection of viral particles by electron microscopy. However, symptoms of PML may be similar to those observed during lymphoma progression into the central nervous system (CNS).
Here we report the case of a patient with diffuse large B-cell lymphoma (DLBCL) treated with R-CHOP who developed clinical signs indicating PML. Intravital diagnostic methods failed to yield an unequivocal diagnosis of PML or lymphoma progression in the CNS. However, a post-mortem examination of brain biopsy specimens performed by electron microscopy demonstrated lesions typical for PML and the presence of viral particles. In addition, immunohistochemical assays identified a massive infiltration of lymphoma cells. The case thus suggests either the extremely rare coexistence of two complications: lymphoma CNS infiltration and PML or induction structural CNS lesions by lymphoma infiltration indistinguishable from PML. The presented findings thus highlight the need for a further review of the current diagnostic criteria for PML.
Author Contributions
Academic Editor: Krzysztof Roszkowski, Department of Radiotherapy, F. Lukaszczyk Oncology Center, Poland.
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2014 Monika, et al.
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Introduction
Progressive multifocal leukoencephalopathy (PML) is a very rare complication associated with rituximab (R) administration. The first two cases of PML were reported in 2002, however since others also emerged, an additional health warning was included in the summary of R characteristics.1, 2 The incidence of PML is estimated to be ca. 2-2.89 cases per 100,000 patients treated with R per year.3
The underlying physiology of PML has not been fully elucidated. It has been proposed that the development of the disease is a consequence of reactivation of JC polyomavirus in the brain cells, causing their damage and ultimately, in the majority of cases, leading to patient death. JC polyomavirus is a DNA virus first isolated in 1971 from the brain of a patient with the initials J.C. (John Cunningham) suffering from Hodgkin’s lymphoma who died of PML. Anti-JCV antibodies are detected in 66-92% of the adult population, with infection occurring mostly in early childhood. Despite of high JC infection prevalence the incidence of PML is low. The disease usually accompanies conditions involving immunosuppression; either associated with the treatment modality or with the existing disease, mainly in HIV infected patients.
Prior to the AIDS epidemic, only ca. 230 cases of PML were documented worldwide. After the advent of AIDS, however, the number of cases of PML increased, not only as a consequence of the growth in the numbers of HIV-positive patients but also due to the emergence of new immunomodulatory drugs (rituximab, natalizumab, efalizumab and others), which highlighted the key role of malfunctions of the immune system in the development of PML. JCV in its latent form has been reported to reside in the kidneys, tonsils, peripheral blood leukocytes and, importantly, also in the brain of healthy individuals who do not exhibit PML symptoms.4 The virus exists in two forms: archetypal type, persisting in the kidneys, and PML JCV type, found exclusively in the brain tissue and peripheral blood lymphocytes, endowed with the potential to destroy glial cells and causing PML.
The diagnostics and diagnosis of PML in patients with B-cell lymphomas treated with immunochemotherapy poses a great challenge to clinicians – especially in view of the fact that clinical symptoms of PML may be similar to those observed during lymphoma progression into the brain. The case study reported here presents a patient with diffuse large B-cell lymphoma (DLBCL), treated with R-CHOP, who developed subtle yet gradually worsening personality changes prompting the initiation of diagnostic procedures. Magnetic resonance imaging (MRI) findings were highly suggestive of PML. Several additional tests performed as part of differential diagnostics with DLBCL progression in the CNS did not provide conclusive results for an unequivocal diagnosis before death. Post-mortem examination combined with histopathological tests and electron microscopic examination of material collected immediately after patient death suggests the coexistence of infiltrative parenchymal lesions by DLBCL cells and a presence of viral infection.
Case study
The patient, aged 61, with history only for hypertension, underwent an emergency surgery for gastrointestinal tract perforation accompanied by diffuse peritonitis. Intraoperatively, a bulky tumour was found in the caecum consistent with DLBCL: CD20+;bcl6+;bcl2+;CD3-;CD15-,CD30-;Ki67 in 70% of cells. Computed tomography (CT) showed involvement of lymph node regions on both sides of the diaphragm. The bone marrow was free from lymphoma infiltration. Blood tests revealed markedly elevated LDH activity – 1,209 U/l (normal range: 240-480 U/l), CRP concentration – 97 mg/l (normal range: 0-5) and anaemia – HGB=9.8 g/dl of the microcytic type – MCV=75 fl. Upon admission WHO performance status was 3, IPI scored 4 and the patient was HIV negative.
After corticosteroids pre-treatment R-CHOP was administrated at doses reduced by 25-50%. The patient improved gradually and continued R-CHOP at standard recommended doses outpatient. The patient remained well until the fifth course of R-CHOP when the patient’s wife reported changes in her husband’s behaviour in the form of memory disturbances and subtle changes in personality. During the medical interview the patient denied having any complaints. He responded logically, and was well oriented to place, time and himself. The patient’s consciousness was clear. Physical examination including neurological assessment failed to demonstrate signs of elevated intracranial pressure or any other abnormalities. On subsequent days of observation, however, subtle changes in the patient’s behaviour were identified, encompassing periodic confusion, lack of critical thinking, agitation – without accompanying pathological neurological symptoms. The family reported progressive changes in the patient’s personality. Neurological assessment still showed no significant abnormalities indicating focal lesions. CT demonstrated a nearly complete remission of lymph node lesions. MRI of the brain, however, revealed multiple small diffuse lesions of the infiltrative and oedematous types occupying almost entirely white matter and, to a lesser extent, also the cortex of the frontal lobes and the left temporal lobe, and periventricularwhite matter of the left occipital lobe. Similar abnormalities were also found bilaterally in the deep cerebral structures, in the right thalamus, penetrating into the posterior limb of the internal capsule. Following contrast administration there was moderate enhancement in the region of the most pronounced lesions in the frontal lobes. Additionally, the examination showed the presence of a convexity meningioma located in the right parietal area, measuring 17x12 mm of no clinical significance. No signs of infiltration were found within the meninges. The combined findings suggested either multifocal leukoencephalopathy or progression of the lymphoma.
Flow cytometric characterization of the cerebrospinal fluid (CSF) showed the presence of 41% of lymphoid cells; the remaining cells being macrophages. An analysis of lymphoid cells revealed exclusively T-cells: CD45+; CD5+ and CD3+ and a complete absence of B-cells CD19/20+. A PCR assay failed to identify the DNA of the JC virus. As the clinical picture was not typical for CNS infiltration by lymphoma, PML diagnosis was suggested and symptomatic treatment with antioedema agents and immunoglobulin infusion was started. A temporary improvement in somatic symptoms was noted, however the patient’s mental disturbances exacerbated. Open brain biopsy was decided, however the patient’s condition rapidly deteriorated and soon after the patient died after developing signs of cerebral oedema.
Post-mortem examination demonstrated a complete remission of lymphoma lesions in the chest and abdominal cavity. Macroscopically, the brain appeared swollen, with narrowed cerebral sulci and flattened gyri. The arachnoid showed a slight congestion. The left hemisphere, particularly the frontal and temporal lobes, displayed significant losses of grey matter with a marked disproportion in the volume of white matter in relation to the right hemisphere. The presence of meningioma was confirmed.
Classical microscopic examination of the brain (Figure 1) showed areas of infiltration with large immune cells CD20+ suggesting progression of the underlying disease (specimens were collected from multiple locations within both hemispheres): LMP1(-), Ki67(++) in ca. 45% of cells, CD3(-) – visible multiple small CD3(+) T-cells, visible enlarged oligodendroglial nuclei. PCR examination of the brain for JC virus was negative.
Figure 1. Histopathological findings . (A,B) and immunohistochemical reactions (IHC) (C,D) of diffuse large B-cell lymphoma, not otherwise specified (DLBCL, NOS) with secondary involvement of white matter. A. Infiltration composed of medium-sized lymphoid cells in the brain tissue without marked necrosis and with a focal infiltration around small blood vessels (HE,100x). B. Infiltration consisting of DLBCL, NOS cells around a small blood vessel (HE,200x). C.D. IHC reactions: CD20(+) in the majority of DLBCL, NOS cells surrounding the lumen of a small blood vessel (C.200x) and CD3(+) in few isolated normal T-cells (D.200x).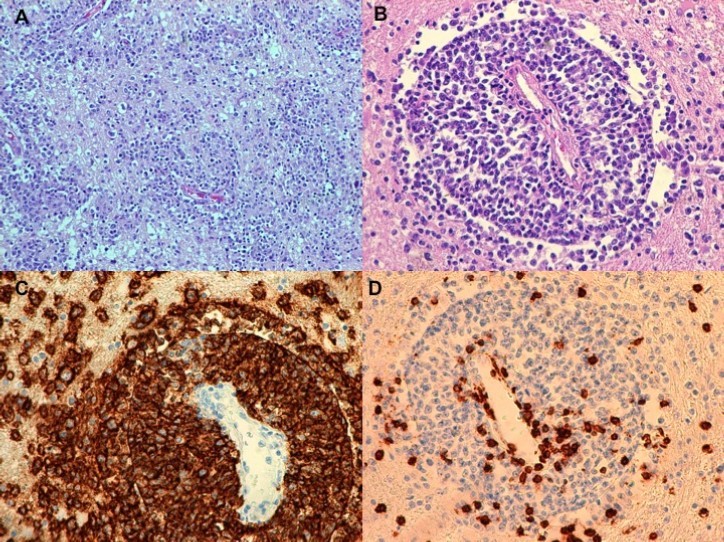
Additional examinations of post-mortem biopsy material taken from subcortical white matter (frontal and temporal lobe regions) by light microscopy LM and transmission electron microscopy TEM were performed. LM analysis of semi-thin sections confirmed the presence of demyelinating foci in the white matter region. Oligodendroglial cells that represent the main site of active JVC infection were found to have structural abnormalities. Particularly notable were enlarged nuclei of these cells and the presence of intranuclear inclusion bodies (Figure 2 and Figure 3). No lymphocytic infiltrations were found in the pathologically altered tissue. Electron microscopy only demonstrated slight post-mortem changes that developed during the 8-hour period between the patient’s death and the fixation of brain tissue. This enhances the value of the TEM results that showed inclusion bodies, i.e. aggregates containing nucleoproteins and virus-like bodies. The structures were located within interchromatin spaces in large oval-shaped oligodendroglial nuclei. The nuclear envelope had numerous isolated scattered enveloped viruses. High microscopic magnification (30-60,000 x) identified an electron-dense centre as viral DNA and an electron-lucent envelope as most likely protein capsid. The viral particles were round in shape, measuring 35-45 nm, and exhibited essentially no variation in shape or size. No fibre (rod) forms were observed. Virus-like particles were also present in the usually narrow strip of cytoplasm, however they were capsid-free.
Figure 2. Additional examinations of post-mortem biopsy material - light microscopy. Subcortical white matter from a patient with PML (routine blue-toluidine staining). Enlarged naked hyperchromatic nucleus of oligodendroglial cell infected with the polyomavirus (arrow) is demonstrated.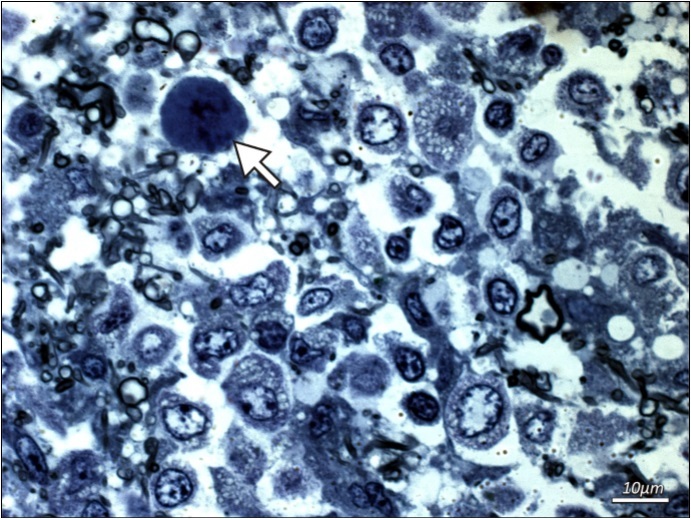
Figure 3. Additional examinations of post-mortem biopsy material - transmission electron microscopy. Electron micrograph presenting virus-like particles randomly distributed in the perinuclear space of infected oligodendrocyte (arrows). Spherical electron-dense centre and electron-lucent viral envelope is visible. The size of the particles, 35 to 45 nm, is typical for JC viruses.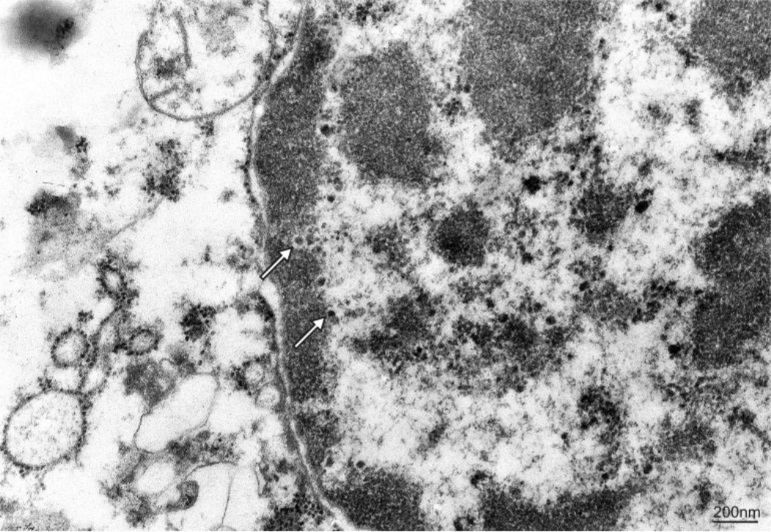
Discussion
Secondary involvement of the brain byDLBCL occurs in ca. 5% of cases. 5 Clinical symptoms observed in our patient were not typical for CNS lymphoma, rather were consistent with PML. Additional arguments favouring PML included location and extent of lesions, MRI findings and absence of lymphoma cells in the CSF. Furthermore, very good response to ongoing treatment was noted, which however does not preclude isolated progression. Reports suggest that CNS progression occurs not only after the completion of cytostatic therapy but also during treatment, also in patients responding outside CNS, as in our case.6
Infiltration of the brain secondary to DLBCL is typically involves the meninges or, less commonly, deep structures of the brain (one third of the cases). In the former scenario, there are meningeal symptoms with severe headaches. In the latter, signs of elevated intracranial pressure (headaches, nausea, vomiting) can be additionally accompanied by changes in behaviour, cranial nerve paralysis, sensory and motor aphasia, convulsions, peripheral neuropathies, balance disorders and even coma. Symptoms identified in our patient were dominated by personality changes, with no signs of elevated intracranial pressure. The patient had no typical risk factors for CNS involvement except of elevated LDH activity.7
The basis for diagnosing CNS involvement byDLBCL is detection of lymphoma cells in the CSF. Currently, flow cytometry offers the highest sensitivity and specificity in detection lymphoma cells. However, in rare cases, in case of deep structures infiltration without meninges involvement, as in our case, CSF is clear of lymphoma cells. Consequently, some authors accept the diagnosis made solely on the basis of existing symptoms and results of imaging studies (preferably MRI with contrast)8, 9, though histopathological diagnosis obtained by stereotactic biopsy is advisable.10 In our case earlier biopsy could have detected lymphoma infiltration and solve the diagnostic dilemma posed by clinical symptoms, MRI findings and lack of lymphoma cells in CSF.
Despite clear evidence of lymphoma infiltration electron microscopy analysis appeared to provide strong evidence for viral inclusions in the brain biopsy specimens and validate the diagnosis of concomitant PML. This is further corroborated by the analysis of semi-thin sections that confirmed the presence of demyelinating foci in the white matter region. The findings were consistent with previously described neurological symptoms. Based on the morphology and size of viral particles, it can be concluded with a high degree of certainty that the case reported here involved viral probably JC infection. The overwhelming majority of oligodendroglial cells displayed ultrastructural signs of degradation: condensation and fragmentation of nuclear chromatin, disintegration of cell membrane, shrinkage of cytoplasm, vacuolization and excessive amounts of free ribosomes indicating infection with the virus. The PML-associated demyelinating process results precisely from oligodendroglial cell death. Electron microscopy revealed marked defects in myelin sheaths. Existing sheaths had a severely disordered arrangement of myelin lamellae, primarily due to delamination.
PML diagnosis itself is a challenging task for clinicians. The consensus statement from the Neuroinfectious Disease Section of the American Academy of Neurology (AAN) released in April 2013 specifies PML diagnostic criteria providing two diagnostic algorithms: one clinical, based on clinical manifestations, imaging tests and laboratory results; and one histopathological, based on histopathological findings. Unequivocal diagnosis of PML through the clinical algorithm requires presence of characteristic symptoms together with imaging findings and JCV DNA identified in the CSF by PCR. Histopathological algorithm requires demonstration of the histopathological triad (demyelination, bizarre astrocytes and enlarged oligodendroglial nuclei) and JCV detection either by immunohistochemistry or electron microscopy, or/and by tissue PCR for JCV11. Other combinations of diagnostic components reduce the likelihood of positive verification of PML diagnosis.
Negative CSF PCR for JCV in the patient discussed in the present study reduces the level of diagnostic certainty for PML. However, following the clinical diagnostic algorithm, negative results of PCR assay of the CSF coupled with positive verification of other diagnostic components yield a probable PML diagnosis (for sure do not exclude it). In line with the histopathological algorithm, negative PCR assay for JCV in the brain does not rule out PML diagnosis, either. What is more, if the remaining two diagnostic criteria are met as in our patient: the presence of triad and viral particles, a definitive diagnosis still can be made. There have been reports of negative CSF PCR in the initial stage of PML, up to three weeks from the emergence of neurological symptoms.12 Some authors claim that a few percent of negative results may in fact be falsely negative.13, 14 In addition, the study by Samorei et al. 15 reports non-uniform distribution of JCV infection in the nervous tissue. Within one organ, there were areas without signs of PML infection (PCR negative) and regions in which the PML process was active to various degrees: from slight to significant infection (PCR positive). In our case the most pronounced abnormalities were found in the vicinity of blood vessels where lymphoma infiltration was present. This observation raises a question of whether lymphoma infiltration could in any way affect the electron microscopy results. However there are no literature reports on such observations.
We conclude that, in the light of the PML diagnostic criteria proposed by the American Academy of Neurology, the findings reported here point quite clearly towards an extremely rare phenomenon of lymphoma progression overlapping with progressive PML. While it cannot be ruled out that a massive lymphoma infiltration in the CNS caused manifestations characteristic for PML – both the typical histopathological triad and the presence of virus-like particles detected by electron microscopy – there are no data suggesting this scenario. If, however, this were the case, PML diagnostic criteria would need to be revised.