Deficiency of Adenosine Deaminase Type 2 (ADA2) DADA2 Masquerade as Lupus
- Open Access
- Peer Reviewed
- Similarity Checked
- CC BY 4.0
Abstract
DADA2 (deficiency of adenosine deaminase type 2) is an autoinflammatory autosomal recessive disease resulting from biallelic loss of function mutations in ADA2 gene. Clinical presentation and age of onset vary widely even among related patients, and variability of symptoms and severity manifestations include bone marrow failure, autoinflammation, immunodeficiency and vasculitis. Here, we report a case of young male with adult onset DADA2, who presented with fever, lower limbs skin rash, joint pain, and anemia resembling systemic lupus erythematous (SLE). DADA2 has an extremely variable clinical phenotype. It was described into three categories: inflammatory/vascular, immune dysregulation, and hematologic. However, the data is scant in describing autoimmunity phenotype in DADA2 and further studies are required to investigate the clinical correlation and presence of autoantibodies. We recommend genetic testing in cases with lupus-like disease especially if there is consanguinity between parents and family history of vasculitis.
Article Information
- Received
- Accepted
- Published
Copyright © 2023 Bayan Almabadi, et al.
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Corresponding author: Correspondence: Bayan Almabadi, Department of Specialized Internal Medicine, King Abdullah Medical City, Makkah, Saudi Arabia —
Competing Interests
The authors have declared that no competing interests
Funding
No specific funding statement was provided by the authors.
Data Availability
No data-availability statement was provided by the authors.
Citation:
Introduction
DADA2 (deficiency of adenosine deaminase type 2) is an autoinflammatory autosomal recessive disease resulting from biallelic loss of function mutations in ADA2 gene. Initially recognized as a syndrome that manifests with fevers, polyarteritis nodosa, livedo racemosa, early-onset stroke, and mild immunodeficiency 1, 2. The clinical phenotype has expanded significantly since the first description of this syndrome in 2014. The estimated prevalence of DADA2 could be as high as 4:100,000 3. Clinical presentation and age of onset vary widely even among related patients 1, 2, 3. Here, we report a case of young male with adult onset DADA2, who presented with fever, lower limbs skin rash, joint pain, and anemia resembling systemic lupus erythematous (SLE).
A 24-year-old male from consanguineous parents, presented with a 1-year history of Raynaud’s phenomenon, repeated epiodes of unexplained fever, arthralgia, morning stiffness of his hands and bilateral lower limb skin rash. He also suffered from dizziness, headache, palpitation, and exertional shortness of breath, and he was found to have anemia of hemoglobin 5.4 mg/dl. He was requiring blood transfusion every two weeks for almost a year. He never had repeated infections apart of herpes zoster twice. He had one sister who was diagnosed with Behcet’s disease in outside facility, she suffered from recurrent mouth ulcers and long standing anemia. Unfortunately, she died at age 27 from ruptured appendix. Upon physical examination of our patient, he looked pale. He had hepatosplenomegaly and palpale axillary lymph nodes. Muskuolskeltal examination did not show any evidence of arthritis. He had maculopapular rash involving his lower limbs bilaterally extending up to his thighs along with hyperpigmentation in his ankle regions Figure 1 A, C. Laboratory workup revealed microcytic hypochromic anemia of hemglobin 5.9 mg/dl without evidence of hemolysis. His Inflammatory markers ESR and CRP were remarkably elevated > 145mm/h and 6.6 mg/dl respectively. His autoimmune workup showed initially negative antinuclear antibodies (ANA) that converted to positive 1:1280, anti-double stranded DNA (anti- dsDNA) was positive 186 IU/ml, rheumatoid factor was positive 53. While antineutrophil cytoplasmic antibodies (ANCA) and lupus anticoagulant were negative. Serum amyloid was elevated 103 (normal range is <= 6.4 mg/dl). CT scan of chest, abdomen and pelvis showed few cervical, axillary, and paratracheal lymphadenopathy with significant hepatosplenomegaly. The patient underwent bone marrow aspiration and biopsy to assess for hematological malignancies explaining his persistent blood-transfuison-dependent anemia which showed pure red cell aplasia. Axillary lymph node biopsy was performed and did not show evidence of lymphoma. Skin biopsy of the maculopapular rash showed perivascular infiltration by neutrophils admixed with eosinophils in the upper dermis, and infiltration of vessel wall by these cells and extravasated red blood cells, these findings were compatible with vasculitis. Although our patient fitted the 2019 European League Against Rheumatism (EULAR) and the American College of Rheumatology (ACR) for SLE, we proceeded for genetic testing given the family history of behcet’s disease and early death, and presence of pure red cell aplasia that is not explainable by lupus. Genetic testing identified two pathogenic variants, c.882-2A>G (Splice acceptor (homozygous) in ADA2. His genetic testing and clinical phenotype confirmed the diagnosis of DADA2. Tumor necrosis factor inhibitors are the mainstay treatment for patients with DADA2 2. Thus, we commenced our patient on adalimumab 40 mg subcutaneous every two weeks. Three months later, his autoinflammatory/vasculitis symptoms; fever, arthralgia, maculopapular rash, and Raynaud’s phenomenon Figure 1 A,B disappeared and his inflammatory markers significantly decreased along with his serum amyloid. Surprisingly, his anti-dsDNA did not improve and continued to increase of 755 IU/ml, anti-TNF therapy. Unfortunately, his anemia also persisted, hemglobin around 5 mg/dl, and he continued to receive blood transfusion every 2 weeks complicated with iron overload. Therefore, he was referred to a bone marrow transplant center for inborn error of immunity and underwent a successful donor-matched haematopoietic stem cell transplantation (HSCT).
Figure 1. Maculopapular rash, biopsy showed evidence of vasculitis (A). After 3 months of initiation of anti-TNF (adalimumab), showed resolution of the maculopapular rash (B). Hyperpigmentation in legs (C)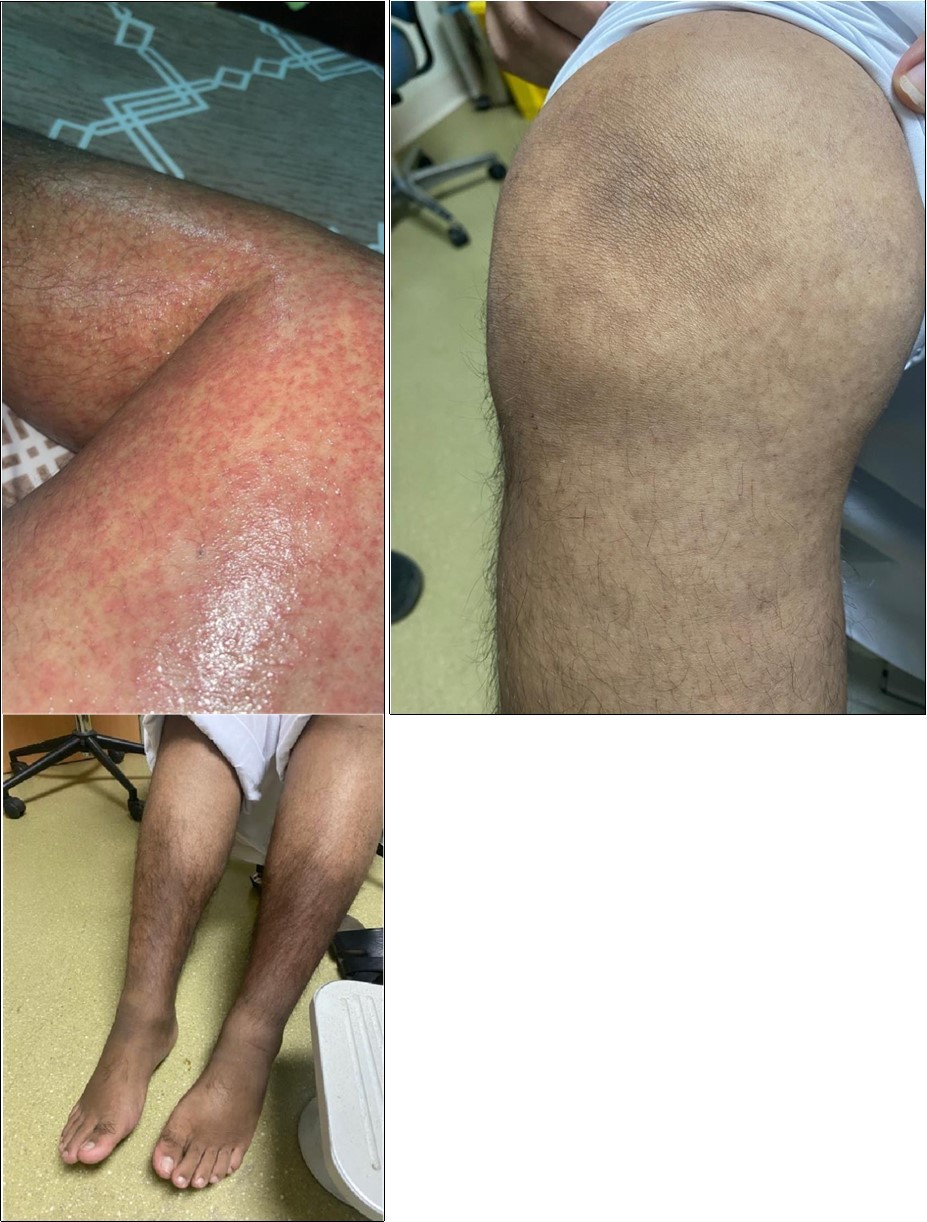
Download figure
Discussion
DADA2 is a rare complex systemic autoinflammatory disorder classified as type-1 interferonpathies as per IUIS classification. It is characterized by reduced or absence of ADA2 enzyme activity, which is an extracellular enzyme that highly expressed in myeloid cells and produced by activated monocytes, macrophages, and dendritic cells. Absent or impaired ADA function causes reduction in deamination of adenosine to deoxyadenosine, and an accumulation of extracellular adenosine leading to abnormal DNA. As a result, dysregulation of NETosis, chronic neutrophil activation, and polarization from the M2 macrophage subtype to the proinflammatory M1 subtype leading to increased inflammatory cytokine production including TNF-α and Interferon (IFN). The net effect, decrease in endothelial cell integrity 3. DADA2 described as a childhood disease with age of onset estimated generally between 5 to 7 years. However, there have also been reports of adult onset, with the oldest patient presenting at age 59 with leg ulceration. 2. Patients with DADA2 can present with three major criterion (1) inflammatory/vascular, (2) immune dysregulation (immunodeficiency, lymphoproliferative disease, and autoimmune manifestations) and (3) hematologic phenotypes. However, most patients presented with significant overlap between these three phenotype groups. Skin is involved in 90% of patients while anemia was only reported in 13% 5. Interestingly, in a cohort from Saudi Arabia hematological and immunological phenotype predominates in these patients 6. Autoimmune features is rare phenotype for patients with ADA2 deficiency. Low titer positive ANA were found in 30% of these patients and less frequent for other autoantibodies such as, anti-ENA, anti-ds DNA, anticardiolipin antibodies, and ANCA 5, 7, 8. Many therapies were used in patients with DADA2 such as glucocorticoids, anti-TNF (etanercept and adalimumab), methotrexate, anakinra, cyclophosphamide, azathioprine, mycophenolate, rituximab, infliximab, and cyclosporine 9. Anti-TNF treatment , is the mainstay of treatment for the inflammatory and vasculopathy phenotypes, it has been shown to be highly efficacious in preventing stroke and significantly reduces stroke risk in DADA2. In addition, it reduces the inflammatory burden (CRP and ESR) , and restores hemoglobin and integrity of endothelial cells in blood vessels 10. However, TNF-α inhibition tend to be less effective in patients with immunodeficiency and hematological phenotypes 9. These patients may be candidates for HSCT. Mortality is high up to 8% of patients before the age of 30 years due to complications of recurrent stroke or infections. HSCT is a definitive cure for DADA2 reversing the refractory cytopenia, vasculopathy and immunodeficiency, returning ADA2 levels to normal and decreasing key cytokines (TNF, IFN, and IL-6) with > 95% survival rate 11.
Conclusion
Given that DADA2 can mimick autoimmune diseases and vasculitis, it should be suspected in patients with vasculitis-like disease and genetic testing is rational here especially if there is a family history of similar presentation and/or early death, or failed commonly used biologic therapy in autoimmune diseases, or additional phenotypes such as bone marrow failure, malignancy and immunodeficiency, because early identification and diagnosis of this disorder has significant treatment implications and can dramatically alter the disease course and HSCT can be life saving in some cases.
References
- 1.Hashem H, Kelly S J, Ganson N J, Hershfield M S.Deficiency of adenosine deami- nase 2 (DADA2), an inherited cause of polyarteritis nodosa and a mimic of other systemic rheumatologic disorders. Curr Rheumatol Rep 2017;19:. 70.
- 2.Meyts I. (2018) Aksentijevich I. Deficiency of Adenosine Deaminase 2 (DADA2): Updates on the Phenotype, Genetics, Pathogenesis, and Treatment. , J Clin Immunol 38(5), 569-578.
- 3.Kendall J L, Springer J M. (2020) The many faces of a monogenic autoinflammatory disease: Adenosine Deaminase 2 Deficiency. Current Rheumatology Reports. 22-64.
- 4.Tangye S, Al-Herz W, Bousfiha A, Cunningham-Rundles C. (2022) Human Inborn Errors of Immunity:. Update on the Classification from the International Union of Immunological Societies expert Committee. Journal of Clinical Immunology. 2022 Oct;42(7): 1473-1507.
- 5.Barron K S, Aksentijevich I, Deuitch N T. (2022) The Spectrum of the Deficiency of Adenosine Deaminase 2: An Observational Analysis of a 60 Patient Cohort. Frontiers in Immunology. 10, 811473-10.
- 6.Alabbas F, Alanzi T.. Genotype and Phenotype of Adenosine Deaminase 2 Deficiency: a Report from Saudi Arabia .J Clin Immunol. 2023 Feb;43(2): 338-349.
- 7.Schepp J, Proietti M, Frede N, Buchta M, Hubscher K et al.Screening of 181 patients with antibody deficiency for deficiency of adenosine deaminase 2 sheds new light on the disease in adulthood. , Arthritis Rheumatol 2017, 1689-700.
- 8.Skrabl-Baumgartner A, Plecko B, Schmidt W M, Konig N, Hershfield M et al.Autoimmune phenotype with type I interferon signature in two brothers with ADA2 deficiency carrying a novel CECR1 mutation. Pediatr Rheumatol. , Online J 2017, 67.
- 9.Ombrello A K, Qin J, Hoffmann P M, Kumar P, Stone D et al.Treatment strategies for deficiency of adenosine deaminase 2. , N Engl J Med 2019, 1582-4.