Drug Design Progress of In silico, In vitro and In vivo Researches
- Open Access
- Peer Reviewed
- Similarity Checked
- CC BY 4.0
Abstract
Drug design, referred to the fields of pharmacology, biotechnology and medicine, is in silico, in vitro and in vivo assay processes of finding new candidate medications based on the biological targets. The in silicoexperiments of drug discovery are involved in the macromolecular structure databases, small molecule databases, molecular docking, de novo drug design and molecular dynamics simulations. The in vitro experiments of drug discovery need evaluate the direct interaction information between ligands and targets as well as the function of ligands on signaling pathway in the cell. The in vivo experiments of drug discovery give the convincing evidence for preclinical trial at the physiological level. In this review, we outline the drug design components of databases, virtual screening tools, biochemical assays, cell-based system and animal models.
Article Information
- Received
- Accepted
- Published
Academic Editor: George Kordas, Twice ERC laureate (Nanotherapy Advanced Grant and PoC Grant) ERC LS7 Panel Member, Member of the ASF 15 Panel
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2018 Qifeng Bai,et al
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Corresponding author: Correspondence: Qifeng Bai, Key Lab of Preclinical Study for New Drugs of Gansu Province, School of Basic Medical Sciences, Lanzhou University, Lanzhou 730000, China —
Competing Interests
The authors have declared that no competing interests exist.
Funding
No specific funding statement was provided by the authors.
Data Availability
No data-availability statement was provided by the authors.
Acknowledgements
The work is supported by the National Natural Science Foundation of China (Grant No. 21605066) and Fundamental Research Funds for the Central Universities (Grant No. lzujbky-2018-92).
Citation:
Introduction
Modern new drug design is the integrated and long-term processes which will cost tens to hundreds of millions dollars from candidate compounds trial to Food and Drug Administration (FDA) approval1. The preclinical drug design trial can be made of in silico, in vitro, and in vivo experiments. The development of information technology and big data accelerate the speed of drug discovery because the high effective and targeted databases are constructed under these circumstances2. The famous databases designed for drug discovery are introduced in detail following review part. Based on the accurate structural models of biological targets and small molecules, the molecular docking software3 can be used to estimate the affinity of ligand in the pocket of biological macromolecules. In addition, the de novo method is another computer-aided design for new drug generation on basis of 3D-structural targets and pharmacophore model4. To study the dynamical interaction between receptors and ligands at the atomic level, the molecular dynamics (MD) simulations supply a reliable and accurate way to explore the binding mechanism between ligands and targets5, 6. Our review describes the popular molecular docking, de novo drug and MD simulation software in drug discovery field. The candidate compounds screened in silico still need be validated to make sure the compound has pharmaceutical activity. The direct binding experiment in vitro between ligands and targets should be trial by using the methods of X-ray crystallography structural analysis, Surface Plasmon Resonance (SPR), etc7. Because the direct binding assay cannot guarantee the activity of screened ligands, the cell signaling pathway response experiment should be performed to check the activity of ligands in cell8. Due to the complex physiological environment, the active ligands in vitro trial may not show any response to the targeting disease in vivo. Hence, it need choose the suitable animal models for in vivo experiments9. In this review, we introduce the database, in silico drug design software, in vitro experiment,and in vivo animal models (see Figure 1). Of course, the sequence of in silico, in vitro and in vivo experiments can be changed according to specific conditions. For instance, if the animal models are easier to be got and cheaper to be bought than biochemical assays, the in vivo experiments can be placed before the in vitro trial. Generally, our review gives the researchers an easy understanding contour for drug design.
Figure 1. The diagram of in silico, in vitro and in vivo for drug design.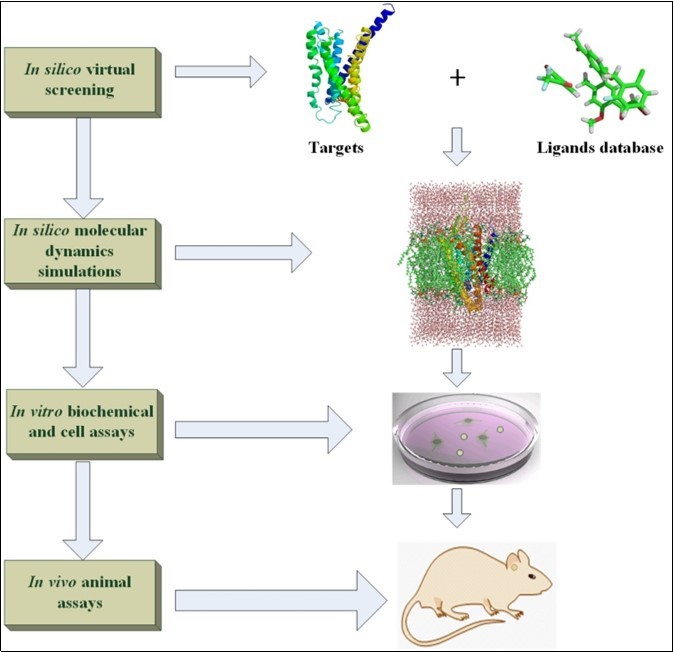
Download figure
In silico Database for Drug Design
With the development of bioinformatics, big data, biology, chemistry and medicine, more and more databases are design to service for the drug discovery10. The databases are divided into macromolecular and small molecular databases. The macromolecular structure databases contain the crystal structures of proteins, nucleic acids, or other biopolymers. Table 1 shows the popular databases of macromolecular crystal structures and theoretical 3D macromolecular structures by homology modeling method. The wwPDB11 currently contains three Protein Data Bank (PDB) databases and one Biological Magnetic Resonance Data Bank which are RCSB PDB, PDBe, PDBj, and BMRB, respectively. The RCSB PDB database is a three dimensional (3D) structural crystallographic database for large biological molecules such as proteins and nucleic acids12 which are collected from NMR spectroscopy, X-ray crystallography, and cryo-electron microscopy13, 14. The databases of PDBe and PDBj lay in Europe and Japan are another two organizations which are responsible for the collection and dissemination of biological macromolecular structures. The Biological Magnetic Resonance Data Bank (BMRB) focuses on the data collection of NMR Spectroscopy from peptides, proteins, nucleic acids, and other biomolecules. Nucleic Acid Database (NDB)15 recruits the functions, structures, analysis, and sequences of experimentally-determined nucleic acids. Molecular Modeling Database (MMDB)16 collects experimentally resolved three-dimensional biomolecule structures under the maintenance of National Center for Biotechnology Information. JenaLib database17 emphasizes the visualization and analysis of three-dimensional biopolymer structures. PDBbind database18, 19 is interested in collecting the experimental binding affinity data. The molecular docking scores can be developed on basis of the collected data of Kd, Ki, and IC50 in PDBbind database. Generally, these macromolecular databases accelerate the drug discovery via providing accuracy and rich structural information, especially, the crystal target data in complex with ligands. Based on the accuracy crystal information, the successful drug screenings and mechanism studies are reported on the targets of transcription factor20, G protein-coupled receptors (GPCRs)21,22,23, transporter24, ion channels receptors25, and so on. IUPHAR/BPS Guide to PHARMACOLOGY26, 27 provides the information of clinical, approved drugs and candidate compounds for the popular targets such as GPCRs, ion channels receptors, kinases, transporters, and so on. It is very simple for users to find the drug comprehensive resources which contain 3D crystal structure targets, relative database links, bioactive ligands from literature, antibodies, functional assays, physiological functions, disease models and so on.
Table 1. Macromolecular structure databases| Structure databases | Description | Weblink |
| wwPDB | Worldwide macromolecular structures | www.wwpdb.org |
| RCSB PDB | Macromolecular structures | www.rcsb.org |
| PDBe | Macromolecular structures in Europe | www.ebi.ac.uk/pdbe |
| PDBj | Macromolecular structures in Japan | pdbj.org |
| BMRB | Macromolecular structures from NMR | www.bmrb.wisc.edu |
| NDB | Nucleic acid structure database | ndbserver.rutgers.edu |
| MMDB | 3D macromolecular structures | www.ncbi.nlm.nih.gov/Structure/MMDB/mmdb.shtml |
| JenaLib | 3D biopolymer structures | jenalib.leibniz-fli.de |
| PDBbind | 3D macromolecular structures in complex with ligands | www.pdbbind.org.cn |
| IUPHAR/BPS Guide to PHARMACOLOGY | Pharmacological targets and substances | www.guidetopharmacology.org |
| ModBase | Theoretical 3D macromolecular structures | modbase.compbio.ucsf.edu/modbase-cgi/index.cgi |
| PMP | Theoretical 3D macromolecular structures | www.proteinmodelportal.org |
| SWISS-MODEL | Theoretical 3D macromolecular structures | swissmodel.expasy.org/repository |
Until now, the number of proteins with crystal structures is limited because there are still some intractably unresolved crystal proteins and nucleic acids structures. The targets without the crystal structures cannot supply the accurate 3D models for drug discovery. In order to screen the candidate compounds based on the active sties of targets without crystal structure, it need predict the 3D structures from the sequence of proteins or nucleic acids. The ModBase28, PMP29, and SWISS-MODEL30 can help the researchers build the 3D theoretical models of targets by comparative modeling methods. The theoretical 3D macromolecular structures also give the reliable 3D models to screen the potential drugs from the large of small molecule database. Some studies have shown homology model can be considered as the valid target to perform the virtual screening31, 32. The crystal and predicted models have been widely applied into the research field of drug discovery.
Drug discovery not only needs the reliable models, but also is relied on huge of small molecules with reasonable conformations. Table 2 shows the popular and special purpose small molecule databases for drug discovery. ZINC database33, 34 is considered as an open-access commercially-available screening library which contains over 35 million purchasable small molecules for virtual screening. ZINC database, which supplies Lead-Like, Drug-Like, Fragment-Like for different research needs, has been reported to be used to screen the candidate compounds by molecular docking methods35, 36. PubChem database currently consists of compounds, substances and bioassay databases which recruit the 93.9 million, 249 million and 1.25 million entries, respectively. Therefore, PubChem37 database can be used to dig out the potential bio-activated compounds through molecular docking and deep learning studies. DrugBank38 is a comprehensive and open-access database containing detail information of drug data and drug targets. In combination with the ZINC database, it has been used to find the ligands via molecular docking based screening39, 40. The databases of ChemBridge, Specs and ChemDiv can supply with the commercially customized screening libraries for the drug development targeting to research receptors such as cyclophilin D41, p-Hydroxyphenylpyruvate dioxygenase (HPPD)42, SETDB1/ESET43, and so on. The e-Drug3D44 database recruits the 1852 molecular structures which approved by Food and Drug Administration (FDA) between 1939 and 2017 with a molecular weight ≤ 2000. The e-Drug3D can be used as the benchmark for finding the effective candidate compounds. Super Natural II45 is a database of natural compounds with physicochemical properties, predicted toxicity information, 2d structures, and vendors. In all, ~75% of FDA approved small molecular drugs are from natural compounds or its derivatives46. Hence, Super Natural II database has a good promising for drug development by in silico method. The Cambridge Structural Database (CSD)47 and Crystallography Open Database (COD)48 are mainly interested in the collection of small molecule organic and inorganic crystal structures. It cannot only be used for drug virtual screening, but also give the accurate structural information for other computing such as quantum chemical calculation. The KEGG Ligand database49 comprises COMPOUND, REACTION, and ENZYME which is responsible for collecting the chemical compounds, chemical reactions, and enzyme molecules, respectively. The nucleic acid ligand database (NALDB)50 and small molecule pathway database (SMPDB)51 are designed for the special purpose of drug discovery. The NALDB provides the detail experimental data of small molecules which target to nucleic acid structures. The SMPDB gives the ligands interactive network on signal pathway found in human which is used to elucidate the drug-action signaling pathways. The MarinChem3D (mc3d.qnlm.ac), which locates at National Laboratory for Marine Science and Technology (Qingdao) in China, publishes more than 30,000 well-defined 3D structures of marine natural products. It can be used to perform the virtual screening studies directly. The MarinChem3D gives a promising way to find candidate ligands targeting to receptors from the ocean.
Table 2. Small molecule databases| Ligand databases | Description | Weblink |
| ZINC | Over 35 million purchasable compounds for virtual screening | zinc.docking.org |
| PubChem | Over 90 million compounds | pubchem.ncbi.nlm.nih.gov |
| DrugBank | More than 11,000 drug entries | www.drugbank.ca |
| Specs | Providing high throughput screening compounds | www.specs.net |
| ChemBridge | Over 1.1 million druglike and leadlike compounds | www.chembridge.com/screening_libraries |
| ChemDiv | Over 1,5 M individual solid screening compounds | www.chemdiv.com/services-menu/screening-libraries |
| e-Drug3D | 1852 FDA approved drugs between 1939 and 2017 | chemoinfo.ipmc.cnrs.fr/MOLDB/index.html |
| Super Natural II | 325,508 natural compounds | bioinf-applied.charite.de/supernatural_new/index.php |
| CSD | Over 900,000 small-molecule organic crystal structures | www.ccdc.cam.ac.uk/solutions/csd-system/components/csd |
| COD | Over 390,000 inorganic crystals and small organic compounds | www.crystallography.net/cod |
| KEGG Ligand | Universe of chemical substances and reactions | www.genome.jp/kegg/ligand.html |
| NALDB | Nucleic acid ligand database | bsbe.iiti.ac.in/bsbe/naldb/HOME.php |
| SMPDB | Over 30,000 small molecule pathways found in humans | smpdb.ca |
| MarinChem3D | Over 30,000 kinds of marine compounds | mc3d.qnlm.ac |
In silico Software for Drug Design
Although the macromolecular structure and small molecule databases are an important factor for drug discovery in silico, it still needs the effective software for performing virtual screening on targets and small molecule databases. In the past decades, various molecular docking software emerges based on different algorithms and molecular formats. Generally, the computational methods for drug discovery can be divided into ligand-based (indirect) and structure-based (direct) techniques52. The ligand-based drug design methods contain quantitative structure-activity relationship (QSAR)53, pharmacophore54, etc. The structure-based drug design contains molecular docking and de novo methods. With the development of genomics and the accumulation of pharmacological information, the big data and deep learning have permeated into the drug discovery fields. For instance, the tensorflow55, which is a deep learning software library, has been used for the drug discovery and molecular dynamics simulations56. These popular computational methods have been integrated into different software for drug design. The QSAR and pharmacophore model can be constructed by Schrödinger or Discovery Studio software. The molecular docking software can be divided into free academic and commercial programs. Besides, molecular dynamics simulations57 are considered as the accurate and dynamical way to study the interaction between targets and ligands. Table 3 shows the free academic, commercial molecular docking programs and molecular dynamics simulations software. LeDock58 is designed based on CHARMM force field parameters by using simulated annealing search algorithm. LeDock shows the very high accuracy in pose prediction and is free for the purpose of academic use. rDock is an open source molecular docking program which can be used to dock ligands into the active sites of proteins and nucleic acids59. AutoDock Vina and AutoDock are two free academic programs for molecular docking. AutoDock can use the flexibility algorithm to dock the ligands into the proteins by Lamarckian genetic algorithm60, 61. AutoDock Vina, which is considered as the new generation of AutoDock, has faster run speed and more accurate binding mode predictions than AutoDock62. UCSF DOCK63 is the first molecular docking program which contains rigid and flexible ligand docking based on the geometric algorithms64. UCSF DOCK can be used to screen the small molecules subset of ZINC database directly. The LigandFit65, Glide66,67,68, GOLD69, MOE Dock and Surflex-Dock70 use the commercial licenses to service for drug virtual screening71,72,73,74,75,76. They show the powerful ability to screen the drugs from small molecule database through checking their citations on Google scholar. Moreover, wang et al. systematically summarize the advantage on the accuracies of binding pose and binding affinity of molecular docking software by comparing with five free academic license programs (LeDock, rDock, AutoDock, AutoDock Vina, and UCSF DOCK) and five commercial license programs (GOLD, LigandFit, MOE Dock, Glide, and Surflex-Dock)77. Furthermore, de novo drug design, which is another computer-aided method for drug discovery, can create the new ligands. LigBuilder78 and MOE Fragment-Based Design (www.chemcomp.com) are the representative de novo drug design software based on fragment linking and growing in the active pocket of targets. The computer-aided software has successfully applied into virtual screening studies and accelerated the process of drug discovery79,80,81. Besides, although the ligands can locate at the active pocket of receptor very well, they may not become the medicine due to the poor ADME (absorption, distribution, metabolism, and excretion)82. ADME can be used to build the computer modeling for the prediction of structure-property relationships and reduce the trial failure of drugs in the clinical phases83. The ADME of drugs can be predicted based on the supported molecular format files by Schrödinger or Discovery Studio software.
Table 3. Software of molecular docking and molecular dynamics simulations| Software | Description | Weblink |
| LeDock | Protein-ligand docking | www.lephar.com/software.htm |
| rDock | Ligands against proteins and nucleic acids | rdock.sourceforge.net |
| AutoDock | Protein-ligand docking | autodock.scripps.edu |
| AutoDock Vina | Protein-ligand docking | vina.scripps.edu |
| UCSF DOCK | Protein-ligand docking | dock.compbio.ucsf.edu |
| LigandFit | Protein-ligand docking | accelrys.com |
| Glide | Protein-ligand docking | www.schrodinger.com |
| GOLD | Protein-ligand docking | www.ccdc.cam.ac.uk/solutions/csd-discovery/components/gold |
| MOE Dock | Protein-ligand docking | www.chemcomp.com |
| Surflex-Dock | Protein-ligand docking | www.jainlab.org |
| Amber | Molecular dynamics simulations | ambermd.org |
| Gromacs | Molecular dynamics simulations | www.gromacs.org |
| NAMD | Molecular dynamics simulations | www.ks.uiuc.edu/Research/namd |
| CHARMM | Molecular dynamics simulations | www.charmm.org |
Molecular docking can perform the high-throughput screening on the huge of small molecule database, while molecular dynamics (MD) simulation is the low-throughput method to evaluate ligand binding pathways84. The software Amber85, Gromacs86, NAMD87, and CHARMM88 are four popular molecular dynamics package mainly designed for simulations of lipids, nucleic acids and proteins (see Table 3). MD software can be used to study the dynamical interaction between targets and ligands at the atomic level. It can profile more detail and accurate interaction information for ligands in the pocket of receptor than molecular docking. Especially, the Poisson–Boltzmann or generalized Born and surface area continuum solvation (MM/PBSA and MM/GBSA) give the good approaches to compute the binding free energy between ligands and biological macromolecules89. As the experiment reported90, MM/GBSA shows the faster and better prediction of binding affinities between ligands and targets than MM/PBSA in the absence of metal. MD simulations have become the popular method to study the mechanism of activated, inactivated states and ligand interaction on different targets, such as GPCRs91,92,93,94,95,96, ion channel receptors97,98,99, etc.
In vitro Biochemical Assays for Compound Screening
One advantage of drug design using high throughput assays and computational tools is that it can largely reduce the use of animals in activity testing. Furthermore, in vitro experiments complemented with computational methods have been extensively used in early drug discovery to select compounds with more favorable ADME and toxicological profiles100,101,102. Most commonly, drugs are organic small molecules produced through chemical synthesis, but biopolymer-based drugs (also known as biopharmaceuticals) produced through biological processes are becoming increasingly more common. In addition, mRNA-based gene silencing technologies may have therapeutic applications.
The predominant strategy used over the last decades consists, first of all, in clarifying the biochemical processes underlying a disease, then identifying an appropriate drug target and finally developing a suitable assay that allows the screening of chemical libraries for small molecules interfering with the target. Based on the various protein targets of diseases, multiple approaches including biological or in silico had been designed to screen new drugs towards the diseases treatment. These approaches are, however, very expensive and demand a great deal of background knowledge. For classical pharmacology, many chemical libraries of synthetic small molecules, natural products or extracts were screened in vitro or in vivo, such as intact cells, whole organisms or cell-free systems to identify substances that have a desirable therapeutic effect.
The early experimental process to approach drug discovery involves several well defined biochemical assays to screen those compounds that can interact or bind with certain binding partners, such as receptor/ligand binding analysis103, 104, enzyme-activity evaluation105, 106. In addition, techniques such as X-ray crystallography structural analysis107, 108, NMR109, 110, calorimetry111, 112, affinity chromatography113, 114, ELISA115,116,117 and protein mass spectrometry118, 119 are common strategic tools in protein-binding studies and play an important role to enhance the structural basis of rational drug design. These techniques aim to detect the separation of compounds from the studied proteins and monitor changes in intrinsic parameters of the targets upon forming a complex with tested drugs. Table 4 shows the biological assays used to detect the binding event between two binding partners, such as protein-protein, protein-nucleic acid, protein-small molecules. In the next, the applications of isothermal titration calorimetry (ITC) and Surface Plasmon Resonance (SPR) are reviewed in detail.
Table 4. Biological assays measuring the binding between macromolecules| Assays | Binding Partners |
| Surface Plasmon Resonance | Protein-protein, protein-DNA, protein-RNA, protein-drug, antibody-antigen, DNA-DNA |
| Enzyme-Linked Immunosorbent Assay | Antibody-antigen, protein-ligand |
| Isothermal Titration Calorimetry | Protein-protein, protein-drug, drug-DNA, protein-DNA, enzyme-substrate |
| Electrophoretic Mobility Shift Assay | Protein-DNA, protein-RNA |
| Thermal shift assay | Protein-drug, enzyme-substrate |
| Protein Fluorescence Quenching | Protein-drug |
| Differential Scanning Calorimetry | Protein-drug |
| Nuclear Magnetic Resonance | Protein-drug |
| Affinity Chromatography | Protein-drug |
| GST Pull-down | Protein-protein |
| Footprinting | Protein-DNA |
| Chromatin Immunoprecipitation | Protein-DNA |
The rapid development of science technology has prompted the emergence of several new approaches. Isothermal titration calorimetry (ITC) is one of the products of the rapid science technology development. It is most often used to investigate the binding of small molecules to larger macromolecules, such as proteins or DNA. In the measurement using ITC, several important parameters involving the binding process can be calculated, including binding affinity, enthalpy changes, and the binding stoichiometry. According to the obtained parameters, the final Gibbs energy changes and entropy changes can be specifically determined120, 121. As ITC gives not only the binding affinity, but also the thermodynamics of the binding interaction, it is typically used as a secondary screening technique in high throughput drug discovery to eliminate false positive hits after primary screening122, 123. Characterization of the binding thermodynamics allows further hit selection and lead optimization as ITC can provide insights into the structure-activity relationship (SAR) for ligand interaction with the target124, 125. Comparing to other techniques such as fluorescence assays and NMR for studying the complex formation, ITC does not need any fluorescent probes or radioactive tags for data analysis. In addition, proteins used in the measurement do not require chemical modification that is ease of use and cost. In spite of various advantages, low throughput, low sensitivity, and large sample requirement are major concerns, which may hamper its application126,127,128.
Another new technique is Surface Plasmon Resonance (SPR), which is the product of nano-science development. The emergence of SPR had greatly reduced the detection limit of biological analysis and it is widely used for the study of ligand binding interactions7, 129, 130. SPR is label-free in that a label molecule is not required for detection of the analytes and capable of measuring real-time quantitative binding affinities and kinetics in sequential binding events131, 132. Moreover, SPR is especially interesting as it can present kinetic information according to affinity data and can be used for thermodynamic studies133,134,135,136. At the same time, SPR biosensor assays can be applied in a wide range of proteins, including membrane proteins, such as G-protein-coupled receptors (GPCRs)137,138,139,140,141. Another application of SPR technology is early ADME (absorption, distribution, metabolism, and excretion) profile prediction for lead compounds in drug discovery trial142. SPR have the merits of real-time measurement, label-free and widespread biomolecules, it is emerging as an essential tool for drug development and has been widely used as a primary screening methodology for drug discovery125, 143, 144. During a SPR measurement, the information includes data about concentration of a binding partner in a mixture as well as kinetic rate constants (association, dissociation rate constants and the equilibrium dissociation constant) for the binding interactions145. Thus, SPR provides insights into the efficacy, safety, duration of action, indication, and patient tolerability of a drug.
Typically, in a compound screening campaign, the selection of evaluation means is highly depended on properties of the target to be studied146. In studies of prion diseases, researchers have proposed several methods to screen new prion inhibitors that would benefit prion-related patients. As prion protein is prone to convert from a dominantly soluble α-helix structure to β-rich insoluble pathogenic aggregates, efforts have been extensively made toward the aggregation dynamic process and discovery new compounds interrupting the aggregation147,148,149,150. Thioflavin T (ThT) can specifically bind to β-rich aggregates accompanying a red shift of the fluorescence emission spectra. Hence, it is frequently used as a dye to monitor the aggregation of prion protein151,152,153,154. Based on this knowledge, Li et al. evaluated the inhibitory effects of several compounds on prion aggregation using ThT as a detector155, 156. Once the aggregation process is interrupted or interfered by a compound, the increase and intensity of ThT fluorescence are delayed or weakened obviously indicating the efficiency of tested compound. Moreover, by combining multiple approaches in drug screening, it would provide more and much accurate information of the drug-target interaction profile. For example, ITC combined with chromatography has been used to identify and isolate unknown target proteins such as receptors or cell/tissue lysates127, 157. Overall, to conduct a successful drug discovery, the evaluation assays should be carefully selected according to different investigating targets.
In vitro Cell-based Approaches
As a common method used in drug discovery, cell-free systems have various advantages, such as fast, microscale and high throughput and the screening results are accurate and stable146, 158, 159. However, the molecular testing models are designed to specific target, which can only provide limited information about the target interactions. Moreover, the effects of certain drugs on an organism are complex and the interactions between two partners are involved in multiple levels that cannot be predicted using biochemical assays. Hence, single molecular high-throughput drug screening technology can no longer meet the needs of new drug discovery today. Then more biologically relevant cell-based screening assays have been developed and are widely used to predict responses of an organism to drugs160, 161. In addition, the cell culture is selected as a model system to predict cellular toxicity, which plays an important role in drug discovery process162.
Tumor cell lines are common cell models used in diseases investigation and drug discovery. Current cell-based models rely heavily on immortalized cell lines, usually derived from human tumors. These models have advantages, such as cost-effective scale up and well consistency. Table 5 displays common human-derived cell lines used in cancer-related investigations. For normal cell lines, they are usually used as controls and toxicology evaluations. Additionally, these cell lines are amenable to genetic engineering, permitting gain and loss of function analysis. While these models demonstrate advantages, they offer limited biological relevance when compared to the intact organ and primary cell types. Currently, primary cells and tissue slices are the gold standards for drug discovery, as they exhibit greater resemblance to the organ of interest163,164,165.
Table 5. Common human cell lines used in several cancer researches| Cancer Types | Normal Cell Lines | Cancer Cell Lines |
| Bladder | SV-HUC-1 | T24, 5637, J82 |
| Breast | DU4475, MCF10A | MCF7, SK-BR-3, HCC38,1590 |
| Colon | - | RKO, CW-2, CBZ, SW48, T84, HRC-6, HT-29 |
| Liver | HL-7702, QSG7701, THLE-3, L-02 | HepG2, Hep3b, HuH-6, Li-7, PLC/PRF/5, HB611, BEL-7404 |
| Lung | MRC-5, HLF-a, HFL1, WI-38, BEAS-2B | A549, NCI-H157, A427, NCI-H524, TKB-1, Lu-165 |
| Pancreas | HPC-Y5 | PANC-1, AsPC-1, HS766T, SW1990 |
| Prostate | WPMY-1, RWPE-1, RWPE-2 | DU145, LNCaP, PC-3, 22RV1, VCaP, 2B4 |
| Renal | HEK-293, HKC, 293FT | SW-13, A498, 786-O, Caki-1, 769-P, UT14 |
Human pluripotent stem cells hold great promise in research and medicine for their unique ability of self-renewing and differentiating to various cell lineages in the body. For different studies, human pluripotent stem cells can be controlled to differentiate to desired cell types to fulfill the investigation purpose166,167,168,169. As general cell lines used in drug screening present limited relevance to the organ of interest, stem cells provide exciting new models and bring new changes for drug discovery and development as well as drug toxicity testing to treat different human diseases170. As we known, it is still a great challenge for drug discovery to neurodegenerative diseases because the biological mechanisms are complex and poorly understood. The lack of models that accurately characterize these dysfunctions blocks further investigations. Fortunately, recent advances in stem cell technology offer researchers available tools to generate human neurons to develop disease resemble assays for small molecules screening. The emergence of adult tissues or cells derived induced pluripotent stem cells (iPSC), which bypass the need for embryos, promotes new investigations of stem cells. Bright et al.171 developed a specific antibody BMS-986168 for the Tau fragment based on human-induced pluripotent stem cells from patients with sporadic Alzheimer’s disease (AD). In 2017, this antibody was licensed by Biogen and entered Phase II clinical trials for AD treatment. Retigabine, another drug under clinical Phase II trial was also derived from iPSC models generated from amyotrophic lateral sclerosis (ALS) patients172. In addition, the recent advances in the production of stem cell-derived hepatocytes and cardiomyocytes combined with cutting-edge engineering technologies supplement the application of stem cells as an attractive alternative model for current drug discovery, which will deliver safer and more efficacious medicines for the patient173,174,175. Moreover, the use of stem cell-derived in vitro systems could reduce animal use and facilitate mechanisms investigation of the toxicants at the same time in toxicity evaluations170. Advancements in pluripotent stem cells and 3D culturing techniques promote the creation of organoids that can accurately recapitulate the properties of various specific subregions of many human organs. Tumor organoids resemble the original tumors much better than cell lines, having a 3D structure, a variety of cells, and similar growth characteristics, they can be applied as a pre-clinical cancer model for drug discovery176, 177. In addition, organoids provide another opportunity to construct cellular models of human diseases that can be used to deeply study the causes of diseases and further identify possible treatment in laboratory178, 179. We believe that advances in stem cell biology would provide more accurate human tissue and disease models for drug development.
In vivo Animal Models
Compared to in vitro screening, in vivo testing is better suited for observing the overall effects of an experiment on a living subject. As in vitro assays can sometimes yield misleading results with drug candidate molecules that are irrelevant in vivo, efficacy verification in vivo is especially crucial in drug discovery process. In addition, whole-organism in vivo screening holds several advantages to small molecule discovery for its target agonistic and holistic.
One of the most important and widely used model organisms in scientific research is zebrafish, which possesses numerous advantages and is the pioneer model for drug screening. It has been used in various research fields, such as gene expression and sequencing180, 181, cancer models182, 183, immune system184 and infectious diseases185, 186. Ongoing research programs have promoted zebrafish model to develop novel therapeutic agents in drug discovery. Drug screens based on zebrafish can not only identify novel classes of compounds with biological effects, but also discover novels uses or targets of existing drugs187. Using a zebrafish screen, a bioactivity from an extract of Jasminum gilgianum plant was discovered to induce the formation of ectopic tailbuds in larvae188. Similarly, some new targets of old medications were identified from zebrafish chemical screening, such as the phenothiazine antipsychotics, which was demonstrated to be toxic to MYC overexpressing thymocytes189. For the commercially available antiangiogenic statin rosuvastatin, a new function was discovered to suppress the growth of prostate cancer in a zebrafish screening of known bioactive compounds190. Other new compound classes were also identified based on zebrafish screens, such as GS4012191 and lenaldekar192.
Another vertebrate animal model frequently used in drug discovery are Xenopus frogs, which belong to the amphibians. As Xenopus frogs share a long evolutionary history with mammals, they are the excellent models to predict human biology. Xenopus have been extensively adopted as a convenient first-line animal model at various stages of drug discovery and development. Since the early 1980s, Xenopus embryogenesis has been much explored and a protocol termed frog embryo teratogenesis assay was applied to identify drugs that pose potential teratogenic hazards, including mortality and malformation193. Embryos and tadpoles were severed as versatile animal models to investigate blood vascular development and angiogenesis. Subsequently, Roland et al.194 uncovered pathways involved in the development of the lymphatic and blood vascular system in Xenopus tadpoles and discovered new compounds and pathways that were not previously known to mediate lymphatic or vascular development. Due to large number and size of the eggs, the rapid development of the embryos and the fact they are amenable to pharmacological, surgical and genetic techniques, Xenopus laevis has been successfully used in searching for embryonic signaling pathways targeting compounds195, particularly the Wnt/β-catenin pathway196, 197.
Despite successful application in drug discovery, the zebrafish and Xenopus models are largely different from the mammals in various aspects including genetics, immune system, and metabolism. Hence, more advanced mammal models were developed to decrease the gap between animal and human diseases, such as dog, rabbit, rat, mouse and the non-human primate Rhesus and Orangutan Monkey. In a research to determine the isolates of pancreatic secretion could be used to treat dogs with diabetes on an animal model dog, promoted the discovery of insulin and then the use in diabetes treatment198. For rabbits used as organism models, they are frequently used to produce antibodies in immunology199. In addition, rabbits are important models to study cardiovascular disease200. Guinea pigs are vertebrate models extensively used by early bacteriologists as hosts for bacterial infections and infectious diseases including viral and parasitic infections201. The mouse is one of the classical model vertebrates and has become the popular choice for developing various in vivo mammalian models as it shares about 85% genome identity to humans and has many physiological systems that are similar to those in humans. In addition, mouse has characteristics such as short life-cycle, techniques for genetic manipulation (inbred strains, stem cell lines, and methods of transformation) and non-specialist living requirements that are predominant and convenient to use. It is commonly used for scientific research in medicine, psychology and genetics202. Comparing to the mouse, the rat has larger size of organs and suborganellar structures and it is particularly used as toxicology models and neurological models203. While for the non-human primate Rhesus and Orangutan Monkeys, they are conventional animal models used in hepatitis, HIV, Parkinson's disease, cognition, and vaccines investigations204, 205. For different scientific research purposes, numerous animal models can be constructed. For example, human tumor xenograft, orthotopic/intratibial tumor models, murine tumor xenograft and patient-derived tumor grafts were built in nude mice or rats to investigate oncology development or screen new cancer drugs206,207,208. Using the constructed models, development of many human tumors including colon cancer, breast cancer, lung cancer, prostate cancer, ovarian cancer, renal cancer, cervical cancer, pancreatic cancer and melanoma has been investigated at different degrees. At the same time, therapeutic compounds targeting these tumors were also discovered either from the existing drugs or from new synthetic compounds.
Despite numerous models developed for drug discovery, most therapeutic drugs still fail in clinical trials. One of the reasons is attributed to sufficient clinical predictive power of our current model systems. Despite the high genetic similarities between human and mice, physiological differences affect the course of diseases in mice models when some genetic disorders in human do not have the same symptoms in mice. In addition, the cell lines and xenografts commonly used are inadequate models that can not highly mimic and accurately predict human diseases. Generally, for a drug discovery research in the laboratory, in combination with the chemical assays, cell-based and in vivo testing would perform more efficiently to obtain effective lead compounds for further drug development. At last, new models are still needed to be developed for scientific researches in the future.
Conclusions
As above review, the entire drug design process can be profiled from in silico, in vitro and in vivo experiments (see Figure 1). Our review summarizes the functions of macromolecular structure databases, small molecule databases, molecular docking software, de novo drug design software, MD simulations software, biochemical assays, cell-based system and animal models. This review shows the detailed individual component of drug design, and gives the comprehensive understanding for the progress of drug design.
References
- 1.Sertkaya A, Wong H H, Jessup A, Beleche T. (2016) Key cost drivers of pharmaceutical clinical trials in the United States. , Clin. Trials 13, 117-126.
- 2.R S Kim, Goossens N, Hoshida Y. (2016) Use of big data in drug development for precision medicine. Expert Rev Precis Med Drug Dev. 1, 245-253.
- 3.N S Pagadala, Syed K, Tuszynski J. (2017) Software for molecular docking: a review. , Biophys Rev 9, 91-102.
- 4.Schneider G, Fechner U. (2005) Computer-based de novo design of drug-like molecules. , Nat. Rev. Drug Discov 4, 649-663.
- 5.Liu X, Shi D, Zhou S, Liu H, Liu H.et al.(2018) Molecular dynamics simulations and novel drug discovery. , Expert Opin Drug Discov 13, 23-37.
- 6.Basith S, Lee Y, Choi S. (2018) Understanding G Protein-Coupled Receptor Allostery via Molecular Dynamics Simulations: Implications for Drug Discovery. Methods Mol. , Biol 1762, 455-472.
- 7.Zeng S, Baillargeat D, Ho H P, K T Yong. (2014) Nanomaterials enhanced surface plasmon resonance for biological and chemical sensing applications. , Chem. Soc. Rev 43, 3426-3452.
- 8.Mazur M, Bujak A, Matloka M, Janowska S, Gunerka P.et al.(2015) Cell-based assay for low- and high-scale screening of the Wnt/beta-catenin signaling modulators. , Anal. Biochem 475, 56-67.
- 9.Daher A, J de Groot. (2018) Rapid identification and validation of novel targeted approaches for Glioblastoma: A combined ex vivo-in vivo pharmaco-omic model. , Exp. Neurol 299, 281-288.
- 11.Berman H, Henrick K, Nakamura H. (2003) Announcing the worldwide Protein Data Bank. , Nat. Struct. Biol 10, 980.
- 12.H M Berman, Westbrook J, Feng Z, Gilliland G, T N Bhat.et al.(2000) The Protein Data Bank. , Nucleic Acids Res 28, 235-242.
- 15.Coimbatore Narayanan B, Westbrook J, Ghosh S, Petrov A I.Sweeney Bet al.(2014) The Nucleic Acid Database: new features and capabilities. , Nucleic Acids Res 42, 114-122.
- 16.Madej T, K J Addess, J H Fong, L Y Geer, R C Geer.et al.(2012) MMDB: 3D structures and macromolecular interactions. , Nucleic Acids Res 40, 461-464.
- 17.Reichert J, Suhnel J. (2002) The IMB Jena Image Library of Biological Macromolecules:. , Nucleic Acids Res 30, 253-254.
- 18.Wang R, Fang X, Lu Y, C Y Yang, Wang S. (2005) The PDBbind database: methodologies and updates. , J. Med. Chem 48, 4111-4119.
- 19.Wang R, Fang X, Lu Y, Wang S. (2004) The PDBbind database: collection of binding affinities for protein-ligand complexes with known three-dimensional structures. , J. Med. Chem 47, 2977-2980.
- 20.Siddiquee K, Zhang S, W C Guida, M A Blaskovich, Greedy B.et al.(2007) Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. , Proc. Natl. Acad. Sci. U. S. A 104, 7391-7396.
- 21.J B Cross. (2018) Methods for Virtual Screening of GPCR Targets: Approaches and Challenges. Methods Mol. , Biol 1705, 233-264.
- 22.Bai Q, Shao Y, Pan D, Zhang Y, Liu H.et al.(2014) Search for beta2 adrenergic receptor ligands by virtual screening via grid computing and investigation of binding modes by docking and molecular dynamics simulations. PLoS One 9. 107837.
- 23.Bai Q, Yao X. (2016) Investigation of allosteric modulation mechanism of metabotropic glutamate receptor 1 by molecular dynamics simulations, free energy and weak interaction analysis. , Sci. Rep 6, 21763.
- 24.Djikic T, Marti Y, Spyrakis F, Lau T, Benedetti P.et al.(2018) Human dopamine transporter: the first implementation of a combined in silico/in vitro approach revealing the substrate and inhibitor specificities. , J Biomol Struct Dyn 1-16.
- 25.Kristam R, S N Rao, A S D’Cruz, Mahadevan V, Viswanadhan V N. (2017) TRPV1 antagonism by piperazinyl-aryl compounds: A Topomer-CoMFA study and its use in virtual screening for identification of novel antagonists. , J. Mol. Graph. Model 72, 112-128.
- 26.S P Alexander, Kelly E, N V Marrion, J A Peters, Faccenda E.. et al.(2017) THE CONCISE GUIDE TO PHARMACOLOGY 2017/18: Overview. Br J Pharmacol 174Suppl1,S1-S16 .
- 27.S D Harding, J L Sharman, Faccenda E, Southan C, A J Pawson.et al.(2018) The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. , Nucleic Acids Res 46, 1091-1106.
- 28.Pieper U, B M Webb, G Q Dong, Schneidman-Duhovny D, Fan H.et al.(2014) ModBase, a database of annotated comparative protein structure models and associated resources. , Nucleic Acids Res 42, 336-346.
- 29.Haas J, Roth S, Arnold K, Kiefer F, Schmidt T.et al.(2013) The Protein Model Portal--a comprehensive resource for protein structure and model information. Database (Oxford)2013,bat031.
- 30.Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G.et al.(2014) SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. , Nucleic Acids Res 42, 252-258.
- 31.A E Leffler, Kuryatov A, H A Zebroski, S R Powell, Filipenko P.et al.(2017) Discovery of peptide ligands through docking and virtual screening at nicotinic acetylcholine receptor homology models. , Proc. Natl. Acad. Sci. U. S. A 114, 8100-8109.
- 32.Feng Z, L V Pearce, Xu X, Yang X, Yang P.et al.(2015) Structural insight into tetrameric hTRPV1 from homology modeling, molecular docking, molecular dynamics simulation, virtual screening, and bioassay validations. , J. Chem. Inf. Model 55, 572-588.
- 33.Irwin J J, Sterling T, Mysinger M M, E S Bolstad, R G Coleman. (2012) ZINC: a free tool to discover chemistry for biology. , J. Chem. Inf. Model 52, 1757-1768.
- 34.J, B K Shoichet. (2005) ZINC--a free database of commercially available compounds for virtual screening. , J. Chem. Inf. Model 45, 177-182.
- 35.Modi P, Patel S, M T Chhabria. (2018) Identification of some novel pyrazolo[1,5-a]pyrimidine derivatives as InhA inhibitors through pharmacophore-based virtual screening and molecular docking. , J Biomol Struct Dyn 1-14.
- 36.Zhang J, Zhu N, Du Y, Bai Q, Chen X.et al.(2015) Dehydrocrenatidine is a novel janus kinase inhibitor. , Mol. Pharmacol 87, 572-581.
- 37.Kim S, P A Thiessen, Bolton E E, Chen J, Fu G.et al.(2016) PubChem Substance and Compound databases. , Nucleic Acids Res 44, 1202-1213.
- 38.D S Wishart, Knox C, A C Guo, Cheng D, Shrivastava S.et al.(2008) DrugBank: a knowledgebase for drugs, drug actions and drug targets. , Nucleic Acids Res 36, 901-906.
- 39.R C Dietrich, L N Alberca, M D Ruiz, Palestro P H, Carrillo C.et al.(2018) Identification of cisapride as new inhibitor of putrescine uptake in Trypanosoma cruzi by combined ligand- and structure-based virtual screening. , Eur. J. Med. Chem 149, 22-29.
- 40.Driessche G Van Den, Fourches D. (2018) Adverse drug reactions triggered by the common HLA-B*57: 01 variant: virtual screening of DrugBank using 3D molecular docking. , J. Cheminform 10, 3.
- 41.Park I, A M Londhe, J W Lim, B G Park, S Y Jung.et al.(2017) Discovery of non-peptidic small molecule inhibitors of cyclophilin D as neuroprotective agents in Abeta-induced mitochondrial dysfunction. , J. Comput. Aided Mol. Des 31, 929-941.
- 42.Fu Y, Y N Sun, K H Yi, M Q Li, H F Cao.et al.(2017) 3D Pharmacophore-Based Virtual Screening and Docking Approaches toward the Discovery of Novel HPPD Inhibitors. , Molecules 22.
- 43.Park I, Y J Hwang, Kim T, Viswanath A N I, A M Londhe.et al.(2017) In silico probing and biological evaluation of SETDB1/ESET-targeted novel compounds that reduce tri-methylated histone H3K9 (H3K9me3) level. , J. Comput. Aided Mol. Des 31, 877-889.
- 44.Pihan E, Colliandre L, J F Guichou, Douguet D. (2012) e-Drug3D: 3D structure collections dedicated to drug repurposing and fragment-based drug design. , Bioinformatics 28, 1540-1541.
- 45.Dunkel M, Fullbeck M, Neumann S, Preissner R. (2006) SuperNatural: a searchable database of available natural compounds. , Nucleic Acids Res 34, 678-683.
- 46.Mohamed A, C H Nguyen, Mamitsuka H. (2016) Current status and prospects of computational resources for natural product dereplication: a review. , Brief Bioinform 17, 309-321.
- 47.C R Groom, I J Bruno, M P Lightfoot, S C Ward. (2016) The Cambridge Structural Database. , Acta Crystallogr B Struct Sci Cryst Eng Mater 72, 171-179.
- 48.Grazulis S, Daskevic A, Merkys A, Chateigner D, Lutterotti L.et al.(2012) Crystallography Open Database (COD): an open-access collection of crystal structures and platform for world-wide collaboration. , Nucleic Acids Res 40, 420-427.
- 49.Goto S, Okuno Y, Hattori M, Nishioka T, Kanehisa M. (2002) LIGAND: database of chemical compounds and reactions in biological pathways. , Nucleic Acids Res 30, 402-404.
- 50.Mishra Kumar, S, Kumar A. (2016) NALDB: nucleic acid ligand database for small molecules targeting nucleic acid. Database. , (Oxford)
- 51.Jewison T, Su Y, F M Disfany, Liang Y, Knox C.et al.(2014) SMPDB 2.0: big improvements to the Small Molecule Pathway Database. , Nucleic Acids Res 42, 478-484.
- 52.Prathipati P, Dixit A, A K Saxena. (2007) Computer-aided drug design: Integration of structure-based and ligand-based approaches in drug design. , Current Computer-Aided Drug Design 3, 133-148.
- 53.Yousefinejad S, Hemmateenejad B. (2015) Chemometrics tools in QSAR/QSPR studies: A historical perspective. , Chemometrics and Intelligent Laboratory Systems 149, 177-204.
- 54.Wermuth C, Ganellin C, Lindberg P, Mitscher L. (1998) Glossary of terms used in medicinal chemistry (IUPAC Recommendations. , Pure Appl. Chem 70, 1129-1143.
- 55.Abadi M, Barham P, Chen J, Chen Z, Davis A.et al.(2016) Tensorflow: a system for large-scale machine learning.OSDI. 265-283.
- 56.Yao K, J E Herr, D W Toth, Mckintyre R, Parkhill J. (2018) The TensorMol-0.1 model chemistry: a neural network augmented with long-range physics. , Chemical science 9, 2261-2269.
- 57.B J Alder, T E Wainwright. (1959) Studies in Molecular Dynamics. , I. General Method. The Journal of Chemical Physics 31, 459-466.
- 58.Zhang N, Zhao H. (2016) Enriching screening libraries with bioactive fragment space. , Bioorg. Med. Chem. Lett 26, 3594-3597.
- 59.Ruiz-Carmona S, Alvarez-Garcia D, Foloppe N, A B Garmendia-Doval, Juhos S.et al.(2014) rDock: a fast, versatile and open source program for docking ligands to proteins and nucleic acids. , PLoS Comput. Biol 10, 1003571.
- 60.G M Morris, D S Goodsell, R S Halliday, Huey R, W E Hart.et al.(1998) Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. , J. Comput. Chem 19, 1639-1662.
- 61.G M Morris, Huey R, Lindstrom W, M F Sanner, R K Belew.et al.(2009) AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. , J. Comput. Chem 30, 2785-2791.
- 62.Trott O, A J Olson. (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. , J. Comput. Chem 31, 455-461.
- 63.P T Lang, S R Brozell, Mukherjee S, E F Pettersen, E C Meng.et al.(2009) DOCK 6: combining techniques to model RNA-small molecule complexes. , RNA 15, 1219-1230.
- 64.I D Kuntz, J M Blaney, S J Oatley, Langridge R, T E Ferrin. (1982) A geometric approach to macromolecule-ligand interactions. , J. Mol. Biol 161, 269-288.
- 65.C M Venkatachalam, Jiang X, Oldfield T, Waldman M. (2003) LigandFit: a novel method for the shape-directed rapid docking of ligands to protein active sites. , J Mol Graph Model 21, 289-307.
- 66.R A Friesner, R B Murphy, M P Repasky, L, J R Greenwood.et al.(2006) Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. , J. Med. Chem 49, 6177-6196.
- 67.T A Halgren, R B Murphy, R A Friesner, H S Beard, L.et al.(2004) Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. , J. Med. Chem 47, 1750-1759.
- 68.R A Friesner, J L Banks, R B Murphy, T A Halgren, Klicic J J.et al.(2004) Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. , J. Med. Chem 47, 1739-1749.
- 69.Jones G, Willett P, R C Glen, A R Leach, Taylor R. (1997) Development and validation of a genetic algorithm for flexible docking. , J. Mol. Biol 267, 727-748.
- 70.A N Jain. (2003) Surflex: Fully automatic flexible molecular docking using a molecular similarity-based search engine. , J. Med. Chem 46, 499-511.
- 71.Lather A, Sharma S, Khatkar A. (2018) Virtual Screening of Novel Glucosamine-6-Phosphate Synthase Inhibitors. , Comb. Chem. High Throughput Screen 21, 182-193.
- 72.Larif S, C B Salem, Hmouda H, Bouraoui K. (2014) In silico screening and study of novel ERK2 inhibitors using 3D QSAR, docking and molecular dynamics. , J. Mol. Graph. Model 53, 1-12.
- 73.Doytchinova I, Atanasova M, Valkova I, Stavrakov G, Philipova I.et al.(2018) Novel hits for acetylcholinesterase inhibition derived by docking-based screening on ZINC database. , J. Enzyme Inhib. Med. Chem 33, 768-776.
- 74.Wang N, Wang L, Xie X Q. (2017) ProSelection: A Novel Algorithm to Select Proper Protein Structure Subsets for in Silico Target Identification and Drug Discovery Research. , J. Chem. Inf. Model 57, 2686-2698.
- 75.Wadood A, Ghufran M, S F Hassan, Khan H.Azam SSet al.(2017) In silico identification of promiscuous scaffolds as potential inhibitors of 1-deoxy-d-xylulose 5-phosphate reductoisomerase for treatment of Falciparum malaria. Pharm. Biol;55,19-32
- 76.Bai Q, Zhang Y, Jin L, Yao X. (2018) Applying Virtual Drug Screening Workflow Module of Schrödinger into College Bioinformatics and Chemoinformatics Teaching. , University Chemistry 33, 66-71.
- 77.Wang Z, H Y Sun, X J Yao, Li D, Xu L.et al.(2016) Comprehensive evaluation of ten docking programs on a diverse set of protein-ligand complexes: the prediction accuracy of sampling power and scoring power. , PCCP 18, 12964-12975.
- 78.Yuan Y X, J F Pei, Lai L H. (2011) LigBuilder 2: A Practical de Novo Drug Design Approach. , J. Chem. Inf. Model 51, 1083-1091.
- 79.R E Amaro, Baudry J, Chodera J, Demir O, J A McCammon.et al.(2018) Ensemble Docking in Drug Discovery. , Biophys. J
- 80.Vilar S, Sobarzo-Sanchez E, Santana L, Uriarte E. (2017) Molecular Docking and Drug Discovery in beta-Adrenergic Receptors. , Curr. Med. Chem 24, 4340-4359.
- 81.Nan J, Du Y, Chen X, Bai Q, Wang Y.et al.(2014) TPCA-1 is a direct dual inhibitor of STAT3 and NF-kappaB and regresses mutant EGFR-associated human non-small cell lung cancers. , Mol. Cancer Ther 13, 617-629.
- 82.Tremaine L, Brian W, DelMonte T, Francke S, Groenen P.et al.(2015) The role of ADME pharmacogenomics in early clinical trials: perspective of the Industry Pharmacogenomics Working Group (I-PWG). , Pharmacogenomics 16, 2055-2067.
- 83.Lombardo F, P V Desai, Arimoto R, K E Desino, Fischer H.et al.(2017) In Silico Absorption, Distribution, Metabolism, Excretion, and Pharmacokinetics (ADME-PK): Utility and Best Practices. An Industry Perspective from the International Consortium for Innovation through Quality in Pharmaceutical Development. , J Med Chem 60, 9097-9113.
- 84.Sledz P, Caflisch A. (2018) Protein structure-based drug design: from docking to molecular dynamics. , Curr. Opin. Struct. Biol 48, 93-102.
- 85.Salomon-Ferrer R, D A Case, R C Walker. (2013) An overview of the Amber biomolecular simulation package. , Wiley Interdisciplinary Reviews-Computational Molecular Science 3, 198-210.
- 86.Spoel D Van Der, Lindahl E, Hess B, Groenhof G, A E Mark.et al.(2005) GROMACS: fast, flexible, and free. , J. Comput. Chem 26, 1701-1718.
- 87.J C Phillips, Braun R, Wang W, Gumbart J, Tajkhorshid E.et al.(2005) Scalable molecular dynamics with NAMD. , J Comput Chem 26, 1781-1802.
- 88.Brooks B R, C L Brooks, 3rd Mackerell AD Jr, Nilsson L.Petrella RJet al.(2009) CHARMM: the biomolecular simulation program. , J Comput Chem 30, 1545-1614.
- 89.Genheden S, Ryde U. (2015) The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. , Expert Opin Drug Discov 10, 449-461.
- 90.Hou T, Wang J, Li Y, Wang W. (2011) Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. , J. Chem. Inf. Model 51, 69-82.
- 91.Bai Q, Zhang Y, Li X, Chen W, Liu H.et al.(2014) Computational study on the interaction between CCR5 and HIV-1 entry inhibitor maraviroc: insight from accelerated molecular dynamics simulation and free energy calculation. , Phys. Chem. Chem. Phys 16, 24332-24338.
- 92.Bai Q, Perez-Sanchez H, Zhang Y, Shao Y, Shi D.et al.(2014) Ligand induced change of beta2 adrenergic receptor from active to inactive conformation and its implication for the closed/open state of the water channel: insight from molecular dynamics simulation, free energy calculation and Markov state model analysis. , Phys. Chem. Chem. Phys 16, 15874-15885.
- 93.Bai Q, Shi D, Zhang Y, Liu H, Yao X. (2014) Exploration of the antagonist CP-376395 escape pathway for the corticotropin-releasing factor receptor 1 by random acceleration molecular dynamics simulations. , Mol. Biosyst 10, 1958-1967.
- 94.Bai Q, Shen Y, Jin N, Liu H, Yao X. (2014) Molecular modeling study on the dynamical structural features of human smoothened receptor and binding mechanism of antagonist LY2940680 by metadynamics simulation and free energy calculation. , Biochim. Biophys. Acta 1840, 2128-2138.
- 95.Bai Q, Zhang Y, Ban Y, Liu H, Yao X. (2013) Computational study on the different ligands induced conformation change of beta2 adrenergic receptor-Gs protein complex. , PLoS One 8, 68138.
- 96.Bai Q, Perez-Sanchez H, Shi Z, Li L, Shi D.et al.(2018) Computational studies on horseshoe shape pocket of human orexin receptor type 2 and boat conformation of suvorexant by molecular dynamics simulations. , Chem Biol Drug Des 92, 1221-1231.
- 97.G H Li. (2018) Exploring conformational states and helical packings in the P2X receptor transmembrane domain by molecular dynamics simulation. , J Biol Phys
- 98.N E Martin, Malik S, Calimet N, J P Changeux, Cecchini M. (2017) Un-gating and allosteric modulation of a pentameric ligand-gated ion channel captured by molecular dynamics. , PLoS Comput Biol 13, 1005784.
- 99.T J Harpole, Delemotte L. (2018) Conformational landscapes of membrane proteins delineated by enhanced sampling molecular dynamics simulations. , Biochim Biophys Acta 1860, 909-926.
- 100.Ripphausen P, Nisius B, Peltason L, Bajorath J. (2010) Quo vadis, virtual screening? A comprehensive survey of prospective applications. , J Med Chem 53, 8461-8467.
- 101.D E Clark. (2008) What has virtual screening ever done for drug discovery?. , Expert Opin Drug Discov 3, 841-851.
- 102.McGregor M J, Luo Z, Jiang X. (2007) screening in drug discovery(Wiley-VCH:. , Weinheim, Germany)Chapter3;25
- 103.E C Hulme, M A Trevethick. (2010) Ligand binding assays at equilibrium: validation and interpretation. , British journal of pharmacology 161, 1219-1237.
- 104.T D Pollard. (2010) A guide to simple and informative binding assays. , Molecular biology of the cell 21, 4061-4067.
- 105.Goddard J-P, Reymond J-L. (2004) Enzyme assays for high-throughput screening. Current opinion in biotechnology 15. 314-322.
- 106.M G Acker, D S Auld. (2014) Considerations for the design and reporting of enzyme assays in high-throughput screening applications. , Perspectives in Science 1, 56-73.
- 107.A M Davis, S J Teague, G J Kleywegt. (2003) Application and limitations of X-ray crystallographic data in structure-based ligand and drug design. , Angew. Chem. Int. Ed. Engl 42, 2718-2736.
- 108.T L Blundell, Jhoti H, Abell C. (2002) High-throughput crystallography for lead discovery in drug design. , Nat. Rev. Drug Discov 1, 45-54.
- 110.Pellecchia M, D S Sem, Wuthrich K. (2002) NMR in drug discovery. , Nat. Rev. Drug Discov 1, 211-219.
- 111.P C Weber, F R Salemme. (2003) Applications of calorimetric methods to drug discovery and the study of protein interactions. , Curr. Opin. Struct. Biol 13, 115-121.
- 112.D A Henriques, J E Ladbury. (2001) Inhibitors to the Src SH2 domain: a lesson in structure--thermodynamic correlation in drug design. , Arch. Biochem. Biophys 390, 158-168.
- 113.G A Ascoli, Domenici E, Bertucci C. (2006) Drug binding to human serum albumin: Abridged review of results obtained with high-performance liquid chromatography and circular dichroism. , Chirality 18, 667-679.
- 114.H S Kim, I W Wainer. (2008) Rapid analysis of the interactions between drugs and human serum albumin (HSA) using high-performance affinity chromatography (HPAC). , Journal of chromatography, B Analyticaltechnologiesinthebiomedicalandlifesciences;870 22-26.
- 115.R M Lequin. (2005) Enzyme immunoassay (EIA)/enzyme-linked immunosorbent assay (ELISA). , Clinical chemistry 51, 2415-2418.
- 116.Alonso N, Guillen R, J W Chambers, Leng F. (2015) A rapid and sensitive high-throughput screening method to identify compounds targeting protein-nucleic acids interactions. Nucleic acids research 43. 52.
- 117.Fung E, Sugianto P, Hsu J, Damoiseaux R, Ganz T.et al.(2013) High-throughput screening of small molecules identifies hepcidin antagonists. , Molecular pharmacology 83, 681-690.
- 118.Glish G L, R W Vachet. (2003) The basics of mass spectrometry in the twenty-first century. Nature reviews. , Drugdiscovery 2, 140-150.
- 119.Mol N J de, M I Catalina, M J Fischer, Broutin I, C S Maier.et al.(2004) Changes in structural dynamics of the Grb2 adaptor protein upon binding of phosphotyrosine ligand to its SH2 domain. Biochimica et biophysica acta 1700. 53-64.
- 120.Pierce M M, C S Raman, B T Nall. (1999) Isothermal Titration Calorimetry of Protein–Protein Interactions. , Methods 19, 213-221.
- 121.Lai M, Lau Y. (2018) Measurement of binding strength between prey proteins interacting with Toxoplasma gondii SAG1 and SAG2 using isothermal titration calorimetry (ITC). , Acta Parasitol 63, 106-113.
- 122.W B Peters, Frasca V, R K Brown. (2009) Recent developments in isothermal titration calorimetry label free screening. , Combinatorial chemistry & high throughput screening 12, 772-790.
- 123.Leavitt S, Freire E. (2001) Direct measurement of protein binding energetics by isothermal titration calorimetry. Current opinion in structural biology. 11, 560-566.
- 124.R W Sarver, Peevers J, W L Cody, F L Ciske, Dyer J.et al.(2007) Binding thermodynamics of substituted diaminopyrimidine renin inhibitors. , Anal. Biochem 360, 30-40.
- 125.Ferenczy G G, G M Keseru. (2010) Thermodynamics guided lead discovery and optimization. Drug Discov. , Today 15, 919-932.
- 126.Liang Y. (2008) Applications of isothermal titration calorimetry in protein science. , Acta Biochim Biophys Sin (Shanghai) 40, 565-576.
- 127.Zhou X, Sun Q, R M Kini, Sivaraman J. (2008) A universal method for fishing target proteins from mixtures of biomolecules using isothermal titration calorimetry. Protein science : a publication of the Protein. , Society 17, 1798-1804.
- 128.J E Ladbury, Klebe G, Freire E. (2010) Adding calorimetric data to decision making in lead discovery: a hot tip. Nature reviews. Drug discovery 9. 23-27.
- 129.Tang Y, Zeng X, Liang J. (2010) Surface Plasmon Resonance: An Introduction to a Surface Spectroscopy Technique. , Journal of chemical education 87, 742-746.
- 130.S G Patching. (2014) Surface plasmon resonance spectroscopy for characterisation of membrane protein–ligand interactions and its potential for drug discovery. , Biochimica et Biophysica Acta (BBA) - Biomembranes 1838, 43-55.
- 131.Rich R L, D G Myszka. (2007) Higher-throughput, label-free, real-time molecular interaction analysis. , Analytical Biochemistry 361, 1-6.
- 132.Saha K, Agasti S S, Kim C, Li X, V M Rotello. (2012) Gold nanoparticles in chemical and biological sensing. , Chem. Rev 112, 2739-2779.
- 133.C F Shuman, P O Markgren, Hamalainen M, U H Danielson. (2003) Elucidation of HIV-1 protease resistance by characterization of interaction kinetics between inhibitors and enzyme variants. , Antiviral research 58, 235-242.
- 134.Gabrielsson J, Dolgos H, P G Gillberg, Bredberg U, Benthem B.et al.(2009) Early integration of pharmacokinetic and dynamic reasoning is essential for optimal development of lead compounds: strategic considerations. Drug discovery today 14. 358-372.
- 135.P O Markgren, Schaal W, Hamalainen M, Karlen A, Hallberg A.et al.(2002) Relationships between structure and interaction kinetics for HIV-1 protease inhibitors. , Journal of medicinal chemistry 45, 5430-5439.
- 136.P S Katsamba, Park S, I A Laird-Offringa. (2002) Kinetic studies of RNA-protein interactions using surface plasmon resonance. , Methods 26, 95-104.
- 137.Navratilova I, A L Hopkins. (2011) Emerging role of surface plasmon resonance in fragment-based drug discovery. Future medicinal chemistry 3. 1809-1820.
- 138.K E Lukong, K W Chang, E W Khandjian, Richard S. (2008) RNA-binding proteins in human genetic disease. Trends in genetics :. , TIG 24, 416-425.
- 139.A J Walhout. (2006) Unraveling transcription regulatory networks by protein-DNA and protein-protein interaction mapping. , Genome research 16, 1445-1454.
- 140.Rich R L, D G Myszka. (2003) A survey of the year 2002 commercial optical biosensor literature. , Journal of molecular recognition : JMR 16, 351-382.
- 141.S H Khan, Farkas K, Kumar R, Ling J. (2012) A versatile method to measure the binding to basic proteins by surface plasmon resonance. , Anal Biochem 421, 385-390.
- 142.Fabini E, U H Danielson. (2017) Monitoring drug-serum protein interactions for early ADME prediction through Surface Plasmon Resonance technology. , Journal of pharmaceutical and biomedical analysis 144, 188-194.
- 143.P J Hajduk, Greer J. (2007) A decade of fragment-based drug design: strategic advances and lessons learned. Nature reviews. , Drugdiscovery 6, 211-219.
- 144.Huber W. (2005) A new strategy for improved secondary screening and lead optimization using high‐resolution SPR characterization of compound–target interactions. , Journal of Molecular Recognition 18, 273-281.
- 145.Minunni M, B A R. (2010) . SPR in Drug Discovery: Searching Bioactive Compounds in Plant Extracts.Ligand-Macromolecular Interactions in DrugDiscovery,(HumanaPress,Totowa,NJ,Totowa), Vol 572 203-218.
- 146.D C Fara, T I Oprea, E R Prossnitz, C G Bologa, B S Edwards.et al.(2006) Integration of virtual and physical screening. Drug Discovery Today:. , Technologies 3, 377-385.
- 147.Bamdad K, Naderi-Manesh H. (2007) Contribution of a putative salt bridge and backbone dynamics in the structural instability of human prion protein upon R208H mutation. , Biochem Biophys Res Commun 364, 719-724.
- 148.Behmard E, Abdolmaleki P, E B Asadabadi, Jahandideh S. (2011) Prevalent mutations of human prion protein: a molecular modeling and molecular dynamics study. , J Biomol Struct Dyn 29, 379-389.
- 149.Chen W, Kamp M W van der, Daggett V. (2010) Diverse Effects on the Native beta-Sheet of the Human Prion Protein Due to Disease-Associated Mutations. , Biochemistry 49, 9874-9881.
- 150.Rossetti G, Giachin G, Legname G, Carloni P. (2010) Structural facets of disease-linked human prion protein mutants: a molecular dynamic study. , Proteins 78, 3270-3280.
- 151.Biancalana M, Koide S. (2010) Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochimica et biophysica acta 1804. 1405-1412.
- 152.O J Rolinski, Amaro M, D J Birch. (2010) Early detection of amyloid aggregation using intrinsic fluorescence. , Biosens Bioelectron 25, 2249-2252.
- 153.Zhou Z, Yan X, Pan K, Chen J, Z S Xie.et al.(2011) Fibril formation of the rabbit/human/bovine prion proteins. , Biophys J 101, 1483-1492.
- 154.Zhou Z, J B Fan, H L Zhu, Shewmaker F, Yan X.et al.(2009) Crowded cell-like environment accelerates the nucleation step of amyloidogenic protein misfolding. , J Biol Chem 284, 30148-30158.
- 155.Li L, Wei W, W J Jia, Zhu Y, Zhang Y.et al.(2017) Discovery of small molecules binding to the normal conformation of prion by combining virtual screening and multiple biological activity evaluation methods. , J Comput Aided Mol Des 31, 1053-1062.
- 156.Li L, Zhu Y, Zhou S, An X, Zhang Y.et al.(2017) Experimental and Theoretical Insights into the Inhibition Mechanism of Prion Fibrillation by Resveratrol and its Derivatives. , ACS Chem. Neurosci 8, 2698-2707.
- 157.Zhou X, R M Kini, Sivaraman J. (2011) Application of isothermal titration calorimetry and column chromatography for identification of biomolecular targets. , Nature 6, 158-165.
- 158.Martis E A R R, Badve R R. (2011) High-throughput screening: The hits and leads of drug discovery—An overview. , Journal of Applied Pharmaceutical Science 1, 2-10.
- 159.T G Fernandes, Diogo M M, D S Clark, J S Dordick, J M Cabral. (2009) High-throughput cellular microarray platforms: applications in drug discovery, toxicology and stem cell research. Trends in biotechnology 27. 342-349.
- 160.Sundberg S A. (2000) High-throughput and ultra-high-throughput screening: solution- and cell-based approaches. Current opinion in biotechnology. 11, 47-53.
- 161.G J Ciambrone, V F Liu, D C Lin, R P McGuinness, G K Leung.et al.(2004) Cellular dielectric spectroscopy: a powerful new approach to label-free cellular analysis. , Journal of biomolecular screening 9, 467-480.
- 162.Szymanski P, Markowicz M, Mikiciuk-Olasik E. (2012) Adaptation of high-throughput screening in drug discovery-toxicological screening tests. , International journal of molecular sciences 13, 427-452.
- 163.Horvath P, Aulner N, Bickle M, A M Davies, E D Nery.et al.(2016) Screening out irrelevant cell-based models of disease. Nature reviews. , Drug discovery 15, 751-769.
- 164.J R Masters, G N Stacey. (2007) Changing medium and passaging cell lines. , Nature 2, 2276-2284.
- 165.C E Nestor, Ottaviano R, Reinhardt D, H A Cruickshanks, H K Mjoseng.et al.(2015) Rapid reprogramming of epigenetic and transcriptional profiles in mammalian culture systems. , Genome biology 16, 11.
- 166.Takahashi K, Yamanaka S. (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. , Cell 126, 663-676.
- 167.Selvaraj V, J M Plane, A J Williams, Deng W. (2010) Switching cell fate: the remarkable rise of induced pluripotent stem cells and lineage reprogramming technologies. Trends in biotechnology 28. 214-223.
- 168.Selvaraj V, Jiang P, Chechneva O, U G Lo, Deng W. (2012) Differentiating human stem cells into neurons and glial cells for neural repair. Frontiers in bioscience 17. 65-89.
- 169.Bai Q, Zhang Y, Jin L, Yao X. (2018) Applying Virtual Drug Screening Workflow Module of Schrödinger into College Bioinformatics and Chemoinformatics Teaching. , University Chemistry 33.
- 170.Liu W, Deng Y, Liu Y, Gong W, Deng W. (2013) Stem cell models for drug discovery and toxicology studies. , Journal of biochemical and molecular toxicology 27, 17-27.
- 171.Bright J, Hussain S, Dang V, Wright S, Cooper B.et al.(2015) Human secreted tau increases amyloid-beta production. , Neurobiol. Aging 36, 693-709.
- 172.B J Wainger, Kiskinis E, Mellin C, Wiskow O, Han S S.et al.(2014) Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. , Cell Rep 7, 1-11.
- 173.M T Donato, Jover R, M J Gomez-Lechon. (2013) Hepatic cell lines for drug hepatotoxicity testing: limitations and strategies to upgrade their metabolic competence by gene engineering. Current drug metabolism 14. 946-968.
- 174.Pradip A, Steel D, Jacobsson S, Holmgren G, Ingelman-Sundberg M.et al.(2016) High Content Analysis of Human Pluripotent Stem Cell Derived Hepatocytes Reveals Drug Induced Steatosis and Phospholipidosis. Stem cells international2016,2475631.
- 175.Takayama K, Mizuguchi H. (2017) Generation of human pluripotent stem cell-derived hepatocyte-like cells for drug toxicity screening. Drug metabolism and pharmacokinetics. 32, 12-20.
- 176.Weeber F, S N Ooft, Dijkstra K K, Voest E E. (2017) Tumor Organoids as a Pre-clinical Cancer Model for Drug Discovery. , Cell Chemical Biology 24, 1092-1100.
- 177.Wetering M van de, Hayley E Francies, Joshua M Francis, Bounova G, Iorio F.et al.(2015) Prospective Derivation of a Living Organoid Biobank of Colorectal Cancer Patients. , Cell 161, 933-945.
- 178.Nakano T, Ando S, Takata N, Kawada M, Muguruma K.et al.(2012) Self-formation of optic cups and storable stratified neural retina from human ESCs. , Cell stem cell 10, 771-785.
- 179.Huch M, Koo B-K. (2015) Modeling mouse and human development using organoid cultures. , Development 142, 3113-3125.
- 180.C B Kimmel, R D Law. (1985) Cell lineage of zebrafish blastomeres. I. Cleavage pattern and cytoplasmic bridges between cells. , Dev. Biol 108, 78-85.
- 181.C B Kimmel, R D Law. (1985) Cell lineage of zebrafish blastomeres. III. Clonal analyses of the blastula and gastrula stages. , Dev. Biol 108, 94-101.
- 183.R M White, Cech J, Ratanasirintrawoot S, Li C Y, P B Rahl.et al.(2011) DHODH modulates transcriptional elongation in the neural crest and melanoma. , Nature 471, 518-522.
- 184.D Le Guyader, M J Redd, Colucci-Guyon E, Murayama E, Kissa K.et al.(2008) Origins and unconventional behavior of neutrophils in developing zebrafish. , Blood 111, 132-141.
- 185.N D Meeker, N S Trede. (2008) Immunology and zebrafish: spawning new models of human disease. , Dev. Comp. Immunol 32, 745-757.
- 186.S A Renshaw, N S Trede. (2012) A model 450 million years in the making: zebrafish and vertebrate immunity. , Dis. Model. Mech 5, 38-47.
- 187.A J Rennekamp, R T Peterson. (2015) 15 years of zebrafish chemical screening. , Curr. Opin. Chem. Biol 24, 58-70.
- 188.Gebruers E, Cordero-Maldonado M L, A I Gray, Clements C, A L Harvey.et al.(2013) A phenotypic screen in zebrafish identifies a novel small-molecule inducer of ectopic tail formation suggestive of alterations in non-canonical Wnt/PCP signaling. , PLoS One 8, 83293.
- 189.Gutierrez A, Pan L, R W Groen, Baleydier F, Kentsis A.et al.(2014) Phenothiazines induce PP2A-mediated apoptosis in T cell acute lymphoblastic leukemia. , J. Clin. Invest 124, 644-655.
- 190.Wang C, Tao W, Wang Y, Bikow J, Lu B.et al.(2010) Rosuvastatin, identified from a zebrafish chemical genetic screen for antiangiogenic compounds, suppresses the growth of prostate cancer. , Eur. Urol 58, 418-426.
- 191.R T Peterson, Shaw S Y, T A Peterson, D J Milan, T P Zhong.et al.(2004) Chemical suppression of a genetic mutation in a zebrafish model of aortic coarctation. , Nat. Biotechnol 22, 595-599.
- 192.Ridges S, W L Heaton, Joshi D, Choi H, Eiring A.et al.(2012) Zebrafish screen identifies novel compound with selective toxicity against leukemia. , Blood 119, 5621-5631.
- 193.J N Dumont, T W Schultz, M V Buchanan, G L Kao. (1983) Frog Embryo Teratogenesis Assay: Xenopus (FETAX) — A Short-Term Assay Applicable to Complex Environmental Mixtures.Short-Term Bioassays in the Analysis of Complex Environmental Mixtures III, eds Waters MD, Sandhu SS,Lewtas J,Claxton L,Chernoff N,Nesnow S(SpringerUS,Boston,MA). 393-405.
- 194.R E Kalin, N E Banziger-Tobler, Detmar M, A W Brandli. (2009) An in vivo chemical library screen in Xenopus tadpoles reveals novel pathways involved in angiogenesis and lymphangiogenesis. , Blood 114, 1110-1122.
- 195.L A Maia, Velloso I, J G Abreu. (2017) Advances in the use of Xenopus for successful drug screening. , Expert Opin Drug Discov 12, 1153-1159.
- 196.G A Nathalia, Barbara F F, Debora Malta C, Alice H R.Alessandro Bolis Costa Set al.(2012) Effects of Natural Compounds on Xenopus Embryogenesis: A Potential Read Out for Functional Drug Discovery Targeting Wnt/β-catenin Signaling. Current Topics in. , Medicinal Chemistry 12, 2103-2113.
- 197.Dominguez I, J B Green. (2000) Dorsal downregulation of GSK3beta by a non-Wnt-like mechanism is an early molecular consequence of cortical rotation in early Xenopus embryos. , Development 127, 861-868.
- 198.D T Karamitsos. (2011) The story of insulin discovery. Diabetes research and clinical practice 93. 2-8.
- 199.Esteves P J d C. (2003) Molecular and population genetic analysis of polymorphism at the antibody loci lgGCH2 and lgVH in lagomorphs.
- 200.S M Pogwizd, D M Bers. (2008) Rabbit models of heart disease. Drug Discovery Today: Disease Models. 5, 185-193.
- 201.D J Padilla-Carlin, D N McMurray, A J Hickey. (2008) The guinea pig as a model of infectious diseases. , Comparative medicine 58, 324-340.
- 202.S E Piret, R V Thakker. (2017) Mouse Models: Approaches to Generate In. Vivo Models for Hereditary Disorders of Mineral and Skeletal Homeostasis.Genetics of Bone Biology and Skeletal Disease (Second Edition), (Elsevier) 89-118.
- 203.Aikawa H, Nonaka I, Woo M, Tsugane T, Esaki K. (1988) Shaking rat Kawasaki (SRK): a new neurological mutant rat in the Wistar strain. , Acta neuropathologica 76, 366-372.
- 204.P E Molina, Amedee A M, Winsauer P, Nelson S, Bagby G.et al.(2015) Behavioral, metabolic, and immune consequences of chronic alcohol or cannabinoids on HIV/AIDs: studies in the non-human primate SIV model. , Journal of Neuroimmune Pharmacology 10, 217-232.
- 205.Zhou Q. (2014) Balancing the welfare: the use of non-human primates in research. , Trends in Genetics 30, 476-478.
- 206.Siolas D, G J Hannon. (2013) Patient-derived tumor xenografts: transforming clinical samples into mouse models. , Cancer research 73, 5315-5319.