
Molecular Cytogenetic Investigations in a Novel Chromosomal Abnormality of t(10;15)(q22;q22) in a Pediatric Precursor-B-Acute Lymphoblastic Leukemia Patient
Abstract
Acute lymphoblastic leukemia (ALL) is a rapid form of leukemia characterized by clonal proliferation and accumulation of immature hematopoietic stem cells of the lymphoid lineage in the bone marrow as well as peripheral blood. Chromosomal aberrations identified in childhood ALL have an important role in disease diagnosis, prognosis and management. We present the results of hematologic, immunophenotypic, cytogenetic, FISH and Multiplex RT-PCR analysis of a 6-year-old boy diagnosed with B-cell precursor Acute Lymphoblastic Leukemia (BCP- ALL). In this study, we identified a novel chromosomal translocation t(10;15)(q22;q22) by cytogenetic and FISH analysis. To the best of our knowledge, this is the first report of this novel chromosomal translocation in this subset of ALL and has not yet been reported elsewhere. This rearrangement may include certain cancer associated tumor suppressor gene(s) or genes involved in apoptosis and transcription regulation, which on loss of normal function may lead to leukaemogenesis.
Author Contributions
Academic Editor: Fernando Luiz Affonso Fonseca, Instituto de Ciências Químicas, Ambientais e Farmacêuticas, Universidade Federal de São Paulo, UNIFESP.
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2014 Prerana Bhandari, et al.
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Introduction:
Acute lymphoblastic leukemia (ALL) is a neoplasm of immature lymphoid progenitors that is mostly of B cell lineage 1. Around 80% of childhood acute leukemia is ALL, especially B cell precursor acute lymphoblastic leukemia (BCP-ALL) (80–85% of ALL) which is the leading cause of cancer-related death in children and young adults 2. Majority of B-ALL cases (approx.75%) exhibit aneuploidy or harbor recurring structural chromosomal rearrangements that are critical initiating events in leukaemogenesis but are insufficient to explain the biology and heterogeneity of disease 3. These rearrangements commonly perturb genes encoding regulators of hematopoiesis, tumor suppressors, oncogenes, or tyrosine kinases but commonly require additional genetic hits to establish the full leukemic phenotype.
An important factor in the diagnosis of ALL is that, karyotype is an independent prognostic indicator with a direct impact on the choice of treatment. The recurrent chromosomal translocations frequently observed in leukemia and lymphoma are important cytogenetic indicators of the genes involved in oncogenesis. The two principal molecular consequences of translocations are the activation of proto-oncogenes and the creation of novel fusion genes [4). Although the former mechanism is frequent among mature B- and T-cell ALL, it is less common in BCP-ALL, in which fusion gene translocations bearing prognostic significance prevail. In this study, we report the first case of a cytogenetically novel pediatric case of Precursor-B-ALL with t(10;15)(q22;q22).
Materials and Methods:
Case History:
A 6-year-old boy was referred to a cancer hospital in August 2012 with chief complaints of low grade fever, petechiae, ecchymosis and bone pain. No evidence of gum hypertrophy or bleeding was noticed at presentation. His complete blood count revealed an anemic condition, with decreased hemoglobin level of 4.5 gm/dL; platelet count 13,000/mm3; and total white blood cell count 7,100 /mm3. Further hematological examination revealed hyper cellular bone marrow with 90% blasts while peripheral blood showed 35% blasts cells. Other clinical finding included presence of lymphadenopathy. Flowcytometric immunophenotyping performed on peripheral blood demonstrated predominant lymphoid blasts cells showing percent positivity as follows: B-Lymphoid markers: CD10-94%; CD19-96%; CD20-16%; CD10/CD19 (co-expression)-94%; CD22-92%; Leucocytic common antigen: CD45-99%; non lineage specific markers:CD34-3%; HLA-DR-97%; myeloid markers: CD13-0%; CD33-0%; CD117-3%; and T-lymphoid markers: CD3-1%, CD5-0% and CD7-0%. These combined pathologic findings from the hematological, bone marrow, and immunophenotyping were suggestive of ALL, and the patient was finally diagnosed with Precursor-B-ALL subtype.
Cytogenetic Analysis:
Conventional cytogenetic analysis was performed on bone marrow aspirate withdrawn at diagnosis as per standard unstimulated direct (0 h, 3 h) and short term (24 h, 48 h) cell culture as per previous reports 5. In brief, cells were cultured in RPMI (Sigma, Schnelldorf, Germany) medium supplemented with 20% FBS (Gibco, Grand Island, NY) at 37°C. After incubation at respective time intervals, the cells were harvested i.e. cell-growth was arrested by exposure to colchicine (HiMedia, 4mg/ml) for 45 minutes, then hypotonised by 0.075 M KCl for 20 minutes, then fixed with chilled Carnoy’s fixative (methanol: glacial acetic acid 3:1). Fixed cells were dropped on chilled frosted slides, then aged overnight at 60°C and stained for GTG banding at resolution of about 400-band level. At least 25 metaphase plates were screened, and 4-5 well spread metaphases were photographed and karyotyped using Ikaros Metasystem Software (Gmbh, Germany). The karyotype was interpreted according to ISCN 2009 6.
Fluorescence in Situ Hybridization (FISH) Analysis:
The FISH analysis was performed from cultured bone marrow cells on metaphase cells using a Vysis LSI probe in order to understand the probable breakpoints involved in this novel rearrangement. The LSI probe was labeled with Spectrum orange specific for 15q22 locus (Abbott Molecular, Des Plaines, IL). The slides were hybridized overnight according to the Vysis protocol and analyzed using Isis Software (Gmbh, Germany).
Molecular Analysis:
In order to rule out the presence of any cryptic, recurrent chromosomal rearrangements such as BCR-ABL, ETV6-RUNX1, TCF3-PBX1, and MLL-AFF1, we did multiplex nested reverse transcriptase PCR as per previous report with minor modification 7. Briefly, total RNA was extracted from the bone marrow aspirate using QIAamp RNA blood mini kit (Qiagen, Hilden, Germany). Fifty nanograms of total RNA were used to prepare cDNA using random hexamer primer (Fermentas, Hanover, MD) and Sensiscript RT Kit (Qiagen). This was followed by two rounds of nested RT-PCR for detection analysis of the fusion genes. Appropriate internal control was also analysed to check the integrity of RNA.
Results:
Cytogenetic Analysis:
Cytogenetic investigations of the bone marrow cells revealed an abnormal karyotype with novel chromosomal rearrangements involving chromosomes 10 and 15, resulting in the karyotype 46,XY,t(10;15)(q22;q22) pattern in all the cells analysed Figure 1. Microscopic evaluation of the metaphase chromosomes revealed reciprocal translocation of a chromosomal segment from the long arm of chromosome 10q22 to the long arm of 15q22, while 15q22 chromosomal segment was translocated to 10q22 thereby demonstrating a novel translocation t(10;15)(q22;q22). Standard phytohemagglutinin-stimulated peripheral blood lymphocyte culture revealed a normal male karyotype, thereby ruling out the possibility of a germ-line origin for this translocation.
Figure 1.(A) Image of a GTG-banded bone marrow metaphase before FISH analysis. (B) Bone marrow metaphase FISH analysis using a LSI probe (red), revealing localization of one red signal on the derivative chromosome 10q22 and other on the normal 15 chromosome (15q22). (C) Representative karyogram of bone marrow cell showing novel translocations t(10;15)(q22;q22).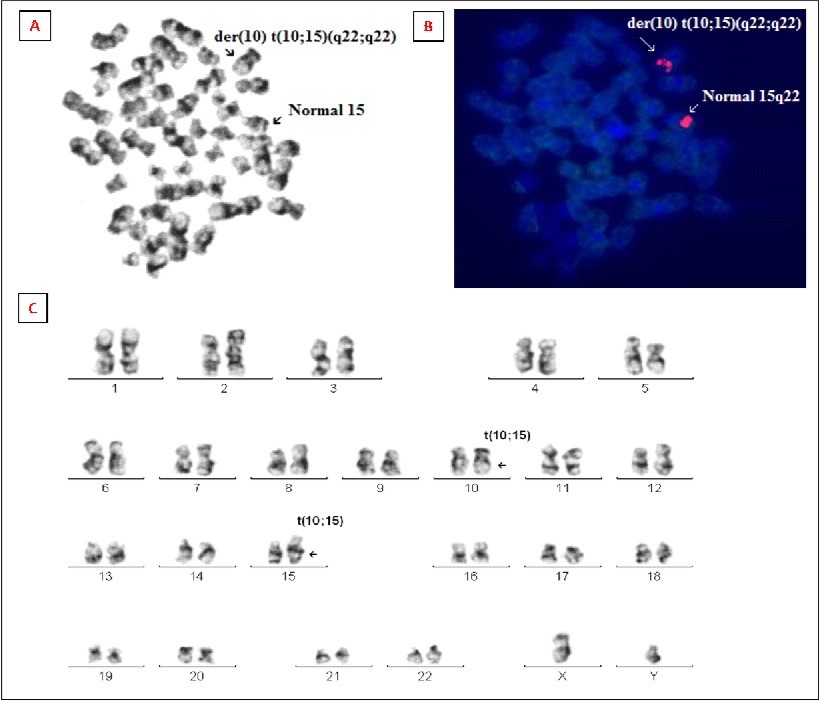
FISH Analysis:
The result of metaphase FISH analysis using the LSI Vysis probe for 15q22 locus is shown in Figure 1. FISH analysis on metaphase cells revealed two orange signals specific to 15q22 locus signal pattern in all the metaphases. Interestingly, out of the two orange signals, one orange signal was located on the long arm of chromosome 10q22 instead of chromosome 15q22 signal absent on the der(15), while the other orange signal was located on normal chromosome 15 thereby confirming presence of rearrangement between 10q22 and 15q22.
Multiplex Nested RT-PCR Analysis:
RT-PCR analysis showed no amplification for the four selected gene fusion transcripts, thereby ruling out the presence of any cryptic rearrangements. Amplification of the internal control was successful suggesting the presence of intact RNA.
Discussion:
The diagnosis of ALL is based on cytomorphology, immunophenotyping, cytogenetic and molecular genetic analysis of the leukemic blast cells in the peripheral blood and bone marrow 8. Analysis of metaphase chromosomes by G-banding is still the gold standard and best method to evaluate chromosomal abnormalities as it provides a global overview of the genome and a baseline to trace the evolution of the disease. The recognition of common genetic alterations in leukemic cells has contributed greatly to our understanding of the pathogenesis and prognosis of ALL 3. The most common prognostic fusion oncogenes in pediatric ALL include BCR-ABL, ETV6-RUNX1, MLL-AF4 and TCF3-PBX1 9. Besides a substantial portion of ALL patients lacking common translocations are characterized by change in their ploidy status (like hypodiploidy, hyperdiploidy etc.], which also serves as an important prognostic factor 10, 11. The observation that up to 25% of pediatric ALL cases lack a recurring chromosomal alteration and the identification of translocation-encoded fusions at birth or in years prior to the onset of leukemia 12 indicates that additional genetic alterations cooperate in leukaemogenesis.
Reported here is a pediatric case of precursor-B-ALL with a novel chromosomal rearrangement t(10;15)(q22;q22) that has not yet been reported in any literature. The initial diagnosis of ALL was made on the basis of clinical features at presentation, high bone marrow cellularity, elevated blast cells in marrow and peripheral blood, low hemoglobin and platelet counts. Flowcytometric immunophenotyping demonstrated predominant lymphoid blasts cells that lacked any myeloid or T-lymphoid markers, while they expressed predominantly B-Lymphoid (CD10, CD19, CD22, HLA-DR and co-expression of CD10 & CD19) markers, which led to the final diagnosis of Precursor- B-ALL subtype. In the present case, we were able to detect t(10;15)(q22;q22) by using a standard cytogenetic technique at 400 band resolution. Microscopic evaluation of leukemic cells revealed that chromosomal segment from the long arm of chromosome 15q22 region was translocated to the distal end on the long arm of chromosome 10q22, resulting in the abnormal karyotype 46,XY,t(10;15)(q22;q22) in all the cells analyzed. Since this abnormality was detected in all the cells, standard phytohemagglutinin-stimulated peripheral blood lymphocyte culture revealed a normal male karyotype, thereby ruling out the possibility of a germ-line origin of this translocation. In order to further understand the breakpoint involvement, we did FISH analysis on metaphase slides using probes located at 15q22. On the basis of FISH results we could ascertain that 15q22 was involved in this rearrangement further confirming our chromosomal findings.
This type of novel chromosomal translocation may entail rearrangements of certain cancer associated tumor suppressor gene/(s) involved in apoptosis and transcription regulation, whose alterations may lead to loss of their normal functions and activation of oncogenic pathways leading to leukemogenesis. As for the actual mechanism or the genes involved in this rearrangement, it still remains unclear. Thus, further molecular characterization of this translocation can unravel the molecular mechanism for disease pathogenesis and will provide further insights into the significance of various gene alterations thereby development of novel targeted therapies.
Acknowledgements
The authors are grateful to the management of SRL Ltd for providing the necessary infrastructure facilities. No other research support is associated with this work.
References
Cited by (3)
This article has been cited by 3 scholarly works according to:
Citing Articles:
Asian Pacific Journal of Cancer Prevention (2015) Crossref
Asian Pacific Journal of Cancer Prevention (2015) OpenAlex
Prerana Bhandari, Firoz Ahmad, R. Dalvi, Neeraja Koppaka, Prajakta Kokate et al. - Asian Pacific Journal of Cancer Prevention (2015) Semantic Scholar
Journal of Hematology and Oncology Research (2014) OpenAlex