
Abstract
Primary thyroid leiomyosarcoma, is extremely rare, with only 19 cases reported in the literature to date. Onset of the tumor, which usually develops in only one lobe of the thyroid, is sudden and the tumor spreads rapidly to surrounding tissues. Preoperative differential diagnosis is extremely difficult. The long-term prognosis for the patient is extremely poor and unrelated to treatment. Indeed, thyroidectomy and neck dissection followed by adjuvant chemotherapy and/or radiation therapy have not been shown to affect rate of recurrence and long-term survival.
The authors describe the case of a patient with leiomyosarcoma of the thyroid gland and review the relevant literature, considering the differential diagnoses and alternative treatment strategies.
Author Contributions
Academic Editor: Zakaria Abd Elmageed, Departments of Urology and Pharmacology School of Medicine, Tulane University, New Orleans LA 70112, USA
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2015 Emilio Mevio, et al.
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Introduction
In the current WHO (2004) classification of thyroid tumors, leiomyosarcoma was included, together with its benign counterpart leiomyoma, among the smooth muscle tumors of the thyroid gland. It is assumed that these tumors arise from degeneration of the smooth muscle of vessel walls of the gland.
Primary thyroid leiomyosarcoma is an extremely rare tumor, of which the literature reports only 19 cases1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 (see Table 1). Approximately 20% of sarcomas involve the head and neck region, affecting mainly the oral, subcutaneous soft tissues, lining of the sinuses and, rarely, the thyroid gland. Leiomyosarcomas account for only 0.014% of primary thyroid tumors. They usually develop in older subjects (mean age of known cases: 66 years) and show no correlation with gender. The development of distant metastases, mainly involving the lungs and liver, is a possibility in thyroid leiomyosarcoma, described in more than 50% of the cases reported in the literature to date (10/19).
Table 1. Thyroid leiomyosarcoma reports in literature| Author | age | sex | T size (cm) | treatment | survival (m) | M1 |
| Mevio 2011 | 80 | M | 8 | Tr+EB+PC | 1 | Y |
| Bertelli 2009 | 39 | M | 3.5 | TT+ND+RT | 48 | N |
| Wang 2008 | 65 | F | 7.5 | TT+ND+CT | 4 | N |
| Uchida 2008 | 65 | F | 4 | Tr+EB+CT | 10 | xxx |
| Mansouri 2008 | 63 | F | 7 | TT | 5 | Y |
| Just 2008 | 83 | F | 9 | PC | 2 | N |
| Day 2007 | 43 | M | 6 | TT+ND+CT | 6 | Y |
| Takayama 2001 | 66 | F | 8.5 | PT+TL | 3 | Y |
| Tsuagawa 1999 | 90 | F | 8 | Tr+EB+PC | 2 | xxx |
| Thompson 1997 | 64 | F | 7.5 | EB | 5 | Y |
| “ | 45 | M | 9 | PT | 11 | Y |
| “ | 68 | M | 1.9 | EB | 18 | Y |
| “ | 83 | M | 5.5 | TT | 3 | Y |
| Ozaki 1997 | 58 | F | 5 | TT+ND | 25 | N |
| Chetty 1993 | 54 | F | 3.5 | PT | 15 | N |
| Iida 1993 | 72 | F | 3 | PT+ND | 51 | Y |
| Kaur 1990 | xxx | xxx | xxx | xxx | 12 | xxx |
| Kawahara 1988 | 82 | M | 5.5 | PT+ND | 4 | N |
| Adachi 1969 | 74 | F | 12 | CT | 1 | Y |
When the tumor is confined to the thyroid the recommended surgical approach is total thyroidectomy with lymph node dissection. When the neoplasm invades surrounding structures, surgical approaches associated with radio and/or chemotherapy have not been found to improve prognosis at all. Survival is variable, ranging from 1 to 51 months, irrespective of the treatment undertaken 1, 8, 10.
Authors Experience
A white male patient, 80 years old, was referred to our hospital with a two-month history of gradual enlargement of the anterior neck. In the previous two weeks he had been experiencing dysphagia (difficult, not painful, swallowing) and dysphonia. He did not present dyspnea. Clinical examination revealed the presence of a hard, but mobile left thyroid lobe mass. Endoscopic fiberoptic examination revealed left vocal fold palsy in adducted position.
Thyroid hormone assay was within normal range.
Chest radiograph and double-contrast examination of the esophagus showed rightward displacement of the trachea and esophagus. Neck ultrasound revealed a massive left thyroid mass, involving superior mediastinal tissue, characterized by confluence of hypo- and hyperechoic areas. Ultrasound-guided fine needle aspiration cytology showed the presence of malignant spindle-shaped cells with moderate atypia. The differential diagnosis included medullary thyroid cancer, anaplastic thyroid cancer and spindle cell sarcoma of either primary or metastatic origin.
Figure 1.CT image of left thyroid lobe tumor causing rightward displacement of the laryngeal structures. The mass has a homogeneous appearance.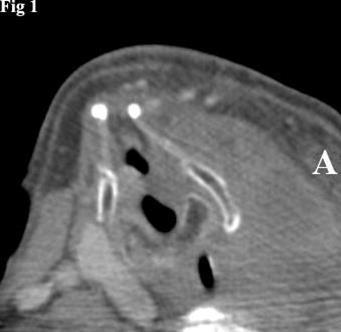
Computed tomography showed a mass, measuring 8x5x7 cm, unevenly distributed in the left thyroid lobe and the presence of colloid cysts and calcified nodules. The mass was causing compression and lateral displacement of the trachea and esophagus with no apparent signs of infiltration of these structures (Figure 1).
There was no evidence of lung lesions. The patient underwent surgery which disclosed a tumor apparently involving only the left lobe of the thyroid. The thyroid capsule was intact.
During surgery, a fresh, left thyroid lobe specimen, consisting of a single fragment of hard, white tissue, 2 cm in diameter, was sent to pathology for intraoperative consultation. The intraoperative consultation diagnosis was: mesenchymal neoplasm, not otherwise specified. The definitive diagnosis was deferred, pending examination of the permanent sections. A total thyroidectomy was planned and attempted, but during surgery it proved impossible to separate the mass from the left trachea and esophagus, to which it was tightly adherent. We decided to remove part of the mass for the purpose of decompression.
Subsequently, a left thyroid lobe specimen consisting of four fragments of hard, white tissue, ranging from 1.8 to 2.7 cm in diameter, were sent to pathology for the final diagnosis.
All submitted fragments were fixed in 10% neutral-buffered formalin, routinely processed and embedded in paraffin. Tissue sections from each fragment were stained with hematoxylin-eosin.
In additional to hematoxylin-eosin the following immunostains were performed: cytokeratins AE1/AE3 (monoclonal M, 1:100, Dako); EMA (M, 1:200, Dako); vimentin (M, 1:75, Dako); actin HHF35 (M, 1:75, Dako); actin 1A4 (M, 1:75, Dako); CEA (M, 1:30, Dako); CD34 (M, 1:30, Dako); TTF-1 (M, 1:100, Leica Biosystems); NSE (M, 1:300, Leica); chromogranin A (M, 1:300, Thermo Scientific); desmin (M, prediluted, Zymed Laboratories Inc.), synaptophysin (M, prediluted, Leica); thyroglobulin, (polyclonal P, 1:7000, Dako); CEA (P, 1:2500, Dako); calcitonin (P, prediluted, Dako).
Figure 2.Hematoxylin and eosin stain showing a fascicle of spindle cells contiguous to residual thyroid follicles. The spindle cells posses blunt-ended, moderately hyperchromatic nuclei. A mitotic figure is evident (top, center).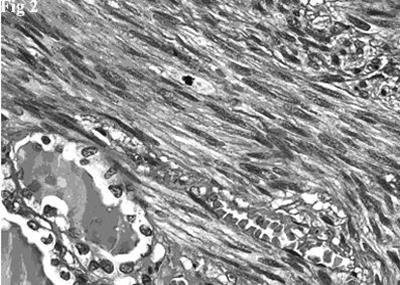
Histologically there was a proliferation of elongated spindle-shaped cells, arranged in interweaving fascicles of varying sizes, intersected at right angles, having blunt-ended, cigar-shaped, centrally located, nuclei. Nuclear hyperchromatism and pleomorphism were generally mild and, occasionally, moderate. The cells had an abundant cytoplasm that varied tinctorially from pink to red in sections stained with hematoxylin-eosin. There was no coagulative tumor cell necrosis. Mitotic figures were easily identified (10 mitoses per 10 HPF). Rare and scattered residual thyroid follicles were surrounded and deformed by the neoplastic fascicles (Figure 2). The follicles, with central colloidal deposits, were lined by flattened or cuboidal epithelial cells.
Figure 3.A. Immunohistochemical stain on the fascicles of tumor cells intersecting each other at right angles was strongly positive for smooth muscle actin. B. Immunohistochemical stain on the spindle tumor cells and on the residual thyroid follicles was positive for EMA.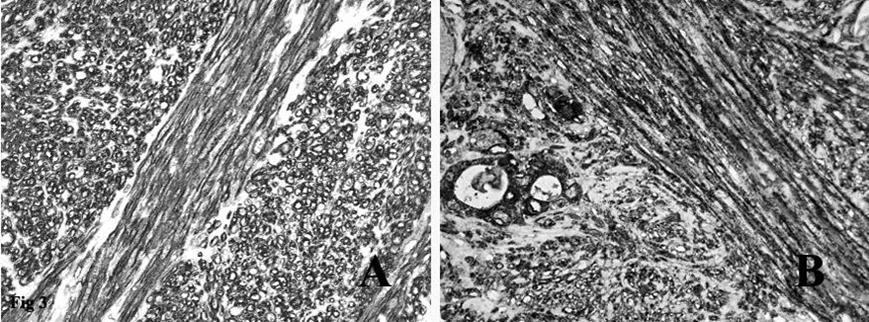
Immunohistochemistry showed that the tumor spindle cells were strongly immunoreactive for vimentin, that is expressed by mesenchymal cells, actin HHF35, actin 1A4 (Figure 3A) and desmin, that are useful markers for the identification of smooth muscle cells. Finally, the spindle cells were positive for EMA (Figure 3B), a representative marker of epithelial cells.
They were negative for the epithelial marker cytokeratin AE1/AE3, a cocktail of two monoclonal antibodies that identifies the majority of human cytokeratins, for thyroid follicular cell markers thyroglobulin and TTF-1, and for neuroendocrine markers chromogranin A, NSE, and synaptophysin. The spindle cells were also negative for S-100, a marker for neural tumors and melanoma, monoclonal and polyclonal CEA, markers expressed by the vast majority of medullary carcinomas, CD 34, which is typically strongly positive in solitary fibrous tumors, and calcitonin.
The entrapped thyroid follicles were positive for cytokeratins, EMA, thyroglobulin and TTF-1, and served as positive internal controls.
On the basis of the clinical, radiographic, histopathological and immunohistochemical features, the final diagnosis was primary thyroid leiomyosarcoma, FNCLCC grade 2.
A week later, the patient presented an acute dyspnea episode associated with cervical tracheal collapse and underwent tracheotomy. A few days after surgery, a CT scan showed evidence of pulmonary micrometastases, not seen on previous imaging studies, and a liver metastasis. In view of the patient’s age, the extent of the primary lesion, the widespread presence of micrometastases and the poor results reported in the literature, we decided to administer only palliative therapy. A month later the patient died.
Discussion and Review of the Literature
Primary leiomyosarcoma of the thyroid gland is a very rare neoplasm typically found in elderly subjects (mean age 66 years). It shows no sex prevalence. In 1969, Adachi et al. reported the first case of primary thyroid metastatic leiomyosarcoma in brain and heart 15. Since then, 19 cases have been reported in the literature, including ours. The etiology of this tumor is unclear. It probably arises from degeneration of smooth muscle cells of the thyroid vessels. It has also been suggested to arise from metaplasia of a previous anaplastic thyroid cancer. The tumor usually develops in only one lobe of the thyroid; it is fast-growing and, due to its tendency to spread to adjacent structures, rapidly leads to the appearance of dysphagia and dysphonia. However, thyroid function is usually preserved.
Primary leiomyosarcoma has no characteristic imaging features that might be useful for diagnostic purposes. Ultrasound-guided fine needle aspiration cytology results do not, as in our case, allow a definitive diagnosis of leiomyosarcoma. It is possible to detect the presence of anaplastic spindle cells, but findings suggesting malignancy do not offer specific guidance as they can also be compatible with other types of cancer: medullary carcinoma, anaplastic carcinoma and sarcoma2, 6.
Histological data are not specific in distinguishing primary from secondary forms. Accordingly, a diagnosis of primary leiomyosarcoma of the thyroid gland must also be based on a thorough clinical diagnostic assessment, which takes into account surgical findings and radiological studies. In our case, clinical and radiological assessment initially revealed only thyroid involvement, thereby demonstrating the primary nature of the tumor. Only subsequently was lung metastasis observed.
Histologically, thyroid leiomyosarcomas must be differentiated from all thyroid epithelial tumors that can show sarcoma-like features and from other sarcomas with spindle cell morphology.
Undifferentiated (anaplastic) thyroid carcinoma (UTC), medullary thyroid carcinoma (MTC), and spindle cell tumor with thymus-like differentiation (SETTLE) are all thyroid epithelial tumors that can mimic sarcomas with a spindle cell pattern.
It can be difficult to exclude a diagnosis of UTC. Histologically, the majority of UTCs are tumors composed of an admixture of spindle cells, pleomorphic giant cells and epithelioid cells. The percentage and distribution of these cell types varies widely from case to case. Tumors composed predominantly or exclusively of spindle cells often have a sarcomatoid appearance. If the tumor cells are arranged in fascicles they can mimic a leiomyosarcoma or a fibrosarcoma.
Immunohistochemical studies play an important role in distinguishing UTCs from leiomyosarcomas. In two large series of UTC patients in which the AE1/AE3 cytokeratin antibody cocktail was used, cytokeratins were detected in about 80% of the cases 16, 17. EMA was less commonly expressed (30-50%) 18. Because some leiomyosarcomas have been shown to contain keratin-positive and/or EMA-positive cells, staining for these epithelial markers cannot be entirely relied upon to distinguish UTCs from leiomyosarcomas. This aberrant immunophenotype in leiomyosarcoma may be a potentially serious diagnostic problem.
In a comprehensive and systematic study, in which immunoreactivity for keratin and EMA was investigated in a large series of 100 leiomyosarcomas, positivity for cytokeratin and EMA was reported in 38% and 44% of the cases, respectively. Although the staining was usually focal, extensive immunoreactivity was observed in 11% with cytokeratin and 6% with EMA 19. In our patient, the finding of diffuse positive myogenic marker staining coupled with absent expression of cytokeratins was a strong indication that the tumor was a leiomyosarcoma and, in view of the above considerations, the diffuse and strong immunoreactivity for EMA does not exclude this diagnosis. Moreover, UTCs do not stain for muscle markers.
The spindle cell variant of medullary carcinoma can mimic a sarcoma. In our patient, the immunohistochemical profile excluded this diagnosis. The fact that the tumor cells lacked positivity for cytokeratin, generic neuroendocrine markers, CEA, TTF-1, and, most important, calcitonin, effectively ruled out the possibility of a MTC.
SETTLE is a very rare malignant thyroid tumor usually found in children, adolescents and young adults. It is a highly cellular tumor which has a lobulated pattern imparted by fibrous septa. Most cases have a biphasic pattern characterized by merging of cytologically bland spindle cells with tubolopapillary epithelial structures. Occasional cases, composed predominantly of spindle cells, may appear monophasic and must be distinguished from leiomyosarcomas.
Unlike SETTLEs, leiomyosarcomas do not have a lobulated pattern and leiomyosarcoma cells usually show much more nuclear hyperchromasia and pleomorphism. Generally, there is far more mitotic activity in leiomyosarcomas than in SETTLEs. Immunohistochemically, the spindle cells in SETTLEs display diffuse immunoreactivity for cytokeratin; in rare cases, they may demonstrate myoepithelial cell differentiation with immunoreactivity for smooth muscle actin and for muscle-specific actin 20. This phenomenon is a potential pitfall in the differential diagnosis. In our case, a diagnosis of SETTLE was excluded on the basis of the absence of a biphasic pattern, cytokeratin expression and a lobulated pattern. Moreover, the patient’s age was not consistent with a SETTLE.
Traditionally, the differential diagnosis of leiomyosarcomas includes other sarcomas that consist of fascicles of moderately differentiated spindle cells such as fibrosarcoma, synovial sarcoma, and malignant peripheral nerve sheath tumor.
Usually, a correct diagnosis can be assigned based on a combination of histologic appearances, patterns of protein expression, assessed by immunohistochemistry, and clinical findings.
Conclusion
In conclusion, primary leiomyosarcoma of the thyroid is a challenging condition requiring close collaboration between radiologist, pathologist and surgeon. Unfortunately, efforts to overcome the difficulties encountered in the differential diagnosis and subsequent therapeutic approaches are undermined by the very poor short-term prognosis linked to the extreme aggressiveness of the tumor.
References
- 1.Bertelli A A, Massarollo L C, Volpi E M, Ueda R Y, Barreto E. (2010) Thyroid gland primary leiomyosarcoma. , Arq Bras Endocrinol Metabol 54, 326-330.
- 2.Wang T S, Ocal I T, Oxley K, Sosa J A. (2008) Primary leiomyosarcoma of the thyroid gland. , Thyroid 18, 425-428.
- 3.Uchida J, Suzaki H. (2008) A case of leiomyosarcoma of the thyroid. , Oto-Rhino-Laryngology Tokyo 51, 145-149.
- 4.Mansouri H, Gaye M, Errihani H, Kettani F, Gueddari B E. (2008) Leiomyosarcoma of the thyroid gland. , Acta Otolaryngol 128, 335-336.
- 5.Just P A, Guillevin R, Capron F, M Le Charpentier, G Le Naour. (2008) An unusual clinical presentation of a rare tumor of the thyroid gland: report on one case of leiomyosarcoma and review of literature. , Ann Diagn Pathol 12, 50-56.
- 6.Day A S, Lou P J, Lin W C, Chou C C. (2007) Over-expression of c-kit in a primary leiomyosarcoma of the thyroid gland Eur Arch Otorhinolaryngol. 264, 705-708.
- 7.Takayama F, Takashima S, Matsuba H, Kobayashi S, Ito N. (2001) MR imaging of primary leiomyosarcoma of the thyroid gland. , Eur J Radiol 37, 36-41.
- 8.Tsugawa K, Koyanagi N, Nakanishi K, Wada H, Tanoue K. (1999) Leiomyosarcoma of the thyroid gland with rapid growth and tracheal obstruction: A partial thyroidectomy and tracheostomy using an ultrasonically activated scalpel can be safely performed with less bleeding. , Eur J Med Res 4, 483-487.
- 9.Thompson L D, Wenig B M, Adair C F, Shmookler B M, Heffess C S. (1997) Primary smooth muscle tumors of the thyroid gland Cancer. 79, 579-587.
- 10.Ozaki O, Sugino K, Mimura T, Ito K, Tamai S. (1997) Primary leiomyosarcoma of the thyroid gland Surg Today. 27, 177-180.
- 11.Chetty R, Clark S P, Dowling J P. (1993) Leiomyosarcoma of the thyroid: immunohistochemical and ultrastructural study Pathology. 25, 203-205.
- 12.Iida Y, Katoh R, Yoshioka M, Oyama T, Kawaoi A. (1993) Primary leiomyosarcoma of the thyroid gland Acta Pathol Jpn. 43, 71-75.
- 13.Kaur A, Jayaram G. (1990) Thyroid tumors: cytomorphology of medullary, clinically anaplastic, and miscellaneous thyroid neoplasms. , Diagn Cytopathol 6, 383-389.
- 14.Kawahara E, Nakanishi I, Terahata S, Ikegaki S. (1988) Leiomyosarcoma of the thyroid gland. A case report with a comparative study of five cases of anaplastic carcinoma. , Cancer 62, 2558-2563.
- 15.Adachi M, Wellmann K F, Garcia R. (1969) Metastatic leiomyosarcoma in brain and heart. , J Pathol 98, 294-296.
- 16.Ordonez N G, Hickey R C, Samaan N A. (1991) Anaplastic thyroid carcinoma. Immunocytochemical study of 32 cases. , Am J Clin Pathol 96, 15-24.
- 17.Venkatesh Y S, Ordonez N G, Schultz P N, Hickey R C, Goepfert H. (1990) Anaplastic carcinoma of the thyroid. A clinicopathologic study of 121 cases. , Cancer 66, 321-330.
- 18.Miettinen M, Franssila K O. (2000) Variable expression of keratins and nearly uniform lack of thyroid transcription factor 1 in thyroid anaplastic carcinoma. , Hum Pathol 31, 1139-1145.
Cited by (2)
- 1.Reddy Bhasker, Aggarwal Vivek, Ajmani Ajay Kumar, Sachan Seema, Khandelwal Deepak, 2019, Primary Leiomyosarcoma of the Thyroid Gland – A Rare Malignancy, European Endocrinology, 15(1), 44, 10.17925/EE.2019.15.1.44
- 2.Wan Mansor Wan Nabila, Abdul Gani Norhaslinda, Abu Dahari Khairul Azlan Shahril, Ahmad Aliza, Japar Jaafar Rohaizam, 2023, Case report: a grievous tale of a rare primary thyroid leiomyosarcoma, The Egyptian Journal of Otolaryngology, 39(1), 10.1186/s43163-022-00364-1