
AB Initio (DFT) and Vibrational Studies of the Synthesized Heterocyclic Compound 2-6-oxo-2-Thioxotetrahydropyrimidin-41H-Ylidene Hydrazine Carbothioamide
Abstract
The structure of the newly synthesized hydrazone derivative 2 - 6 – oxo - 2-thioxotetrahydropyrimidin – 4 1H - ylidene hydrazine carbothioamide (OTTHPYHCT)compound is determined by using spectral information and elemental study. Density functional theory (DFT) studies were performed using the B3LYP/6-31G (d, p) basis set to expand imminent into their structural properties. Frontier molecular orbital (FMO’s) analysis of title compoundwas computed at the same level of theory to get knowledge about their kinetic stability of the molecule by the energy gap value obtained. Global reactivity descriptors are determined to explain the biological activity of the molecule. NBO analysis provides information about charge transfer, delocalization effect, hyperconjugative interactions and the energy responsible for the stabilization of the compound. First hyperpolarizability analysis nonlinear optical response was simulated at the B3LYP/6-31G d, p level of theory as well. Thermodynamic parameters explain vibrational intensity of the molecule.
Author Contributions
Academic Editor: Praveen Kumar Sharma, Lovely Professional University
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2017 N. Kalaiarasi, et al.
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Introduction:
Hydrazones are important organic compounds due to their bioactivity as well as their structural properties. Previous studies have demonstrated that these substances exhibit a wide variety of biological actions. Traces of interest date back to the beginning of the 20th century but in the medicinal field the first report appearing in the fifties as drugs against tuberculosis and leprosy 1, 2. It is known that Hydrazone of thiosemicarbazide compounds possess various pharmacological applications such as antitumor 3, antibacterial, antiviral 4, antiprotozoal and cytotoxic effects 5, 6. They are also used as models in bioinorganic processes 7. Hydrazones posess large pharmacological as well as many biological activities. The effect of the molecular structure on the chemical reactivity has been object of great interest in several disciplines of chemistry and quantum chemistry calculations have been widely used to study the reaction mechanisms and to interpret the experimental results as well as to solve chemical ambiguities. The methods based on the Density Functional Theory (DFT) are established to be very efficient and hence the synthesized compound is briefly discussed using DFT theory and extensive literature survey reveals that quantum chemical calculations have not yet been performed on the title compound both experimentally and theoretically.
Material and Methods:
The chemical used were purchased from Sigma Aldrich Company. Melting points were determined on a Mettler FP51 melting point apparatus and are uncorrected. UV spectra are recorded using ELICO-BL222 spectrophotometer λ max nm using spectral grade methanol as a solvent. Infrared spectra KBr, 4000–400 cm-1 have been recorded on SHIMADZU Fourier transform spectrophotometer. NMR spectra recorded using Bruker 300 MHz spectrometer for 1H-NMR and 100MHz spectrometer for 13C-NMR, with DMSO-d6 as solvent.
Computational Methodology:
Nowadays computational based methods have become very popular to investigate the structure-activity relationships of compounds due to its accuracy and less time consuming. Geometry optimization of the molecules involved in the processes were expertise using the Becke’s three parameters hybrid density functional 8 with the gradient-corrected correlation functional due to Lee, Yang and Parr 9, a combination that gives rise to the well-known B3LYP/6-31Gd,p method 10. Optimized geometries were further used for structural investigations like frontier molecular orbitals analysis FMOs, Molecular electrostatic potential MEP mapping and nonlinear optics NLO properties measurement.
Experimental:
Synthesis of Thiobarbituric Acid TBA
About 6g 0.25mol of sodium metal is dissolved in 200ml of ethanol. To this solution 15g 0.25mol of thiourea and 40ml of diethyl malonate is added. The reaction mixture is refluxed for 6hrs in an oil bath and then vacuum distillated to recover ethanol. The clear solution thus obtained is filtered, cooled in an ice bath overnight and the resulting solution is acidified with HCl. The crude product obtained is collected, washed with 50ml water and dried in oven at 105-1100C for nearly 4 hrs. The obtained white coloured precipitate Thiobarbituric acid-TBA is purified by recrystallization with ethanol. M.P - 243C, yield-80%.
Synthesis of 2 - 6 – oxo - 2-Thioxotetrahydropyrimidin – 4 1H - Ylidene Hydrazine Carbothioamide.
An equimolar mixture of thiobarbituric acid 2.2g 0.01mol, thiosemicarbazide 2.5g 0.01mol are dissolved in ethanol, the content is condensed on an oil bath for 4 hours, then the reaction mixture is standing overnight and the precipitate obtained is filtered washed with ethanol and dried to obtain the Dark yellow colour powdered product. Yield - 68%; M.P. - 1740c. The scheme and of the reaction are represented in Figure 1.
Figure 1.Scheme of The synthetic reaction of formation of 2 - 6 – Oxo - 2- Thioxotetrahydropyrimidin – 4 1H - Ylidene Hydrazine Carbothioamide.
Result and Discussion:
Structural Determination of 2-6-oxo-2-Thioxotetrahydropyrimidin-41H-ylidene Hydrazine Carbothioamide by UV, IR, FT-Raman. 1HNMR, 13CNMR:
The absorption value of λ max at 385.5 nm and 256 nm preferably favors n-π* and n-* transition due to lone pair of atoms present in the compound. The NH frequency value is absorbed experimentally at 3720 cm-1 and 3255 Cm-1 in IR and Raman and theoretically they are assigned at 3570 Cm-1 and 3377 Cm-1. The functional group C=N in the compound confirms the formation of hydrazone and by using DFT/6-31G (d, p) they are computed and the values are probably noted at 1633 Cm-1 which coincides well with experimental IR and FT-Raman values 1639 Cm-1 and 1604 Cm-1. The NH2 and C-N vibrational frequencies are illustrated at 1537 Cm-1 and 1249 Cm-1 in IR and at 1517 Cm-1 and 1301 Cm-1 in Raman which slightly varies from the calculated scaled value 1583 Cm-1 for NH2 functional group and 1308 Cm-1 for C-N group. The lone pair atoms oxygen and sulphur with carbon C=O and C=S shows the vibrations at 1834 Cm-1 and 1165 Cm-1 in IR and in Raman C=S shows a medium peak at 1049 Cm-1 which agrees well with examined values 1763 Cm-1 for C=O and 1127 Cm-1 for C=S. The torsional C=S vibration of the substituted group is absorbed at 644 Cm-1 at IR and 647 Cm-1 at Raman which correlates well with scaled value 612 Cm-1. The frequency output table is shown in Table S1 and the respective IR, Raman theoretical and experimental spectra are shown in Figure 2 and the above spectral values assigned are found to be in good agreement with literature 11.
Figure 2.Experimental and Theoretical IR and Raman spectra of OTTHPYHCT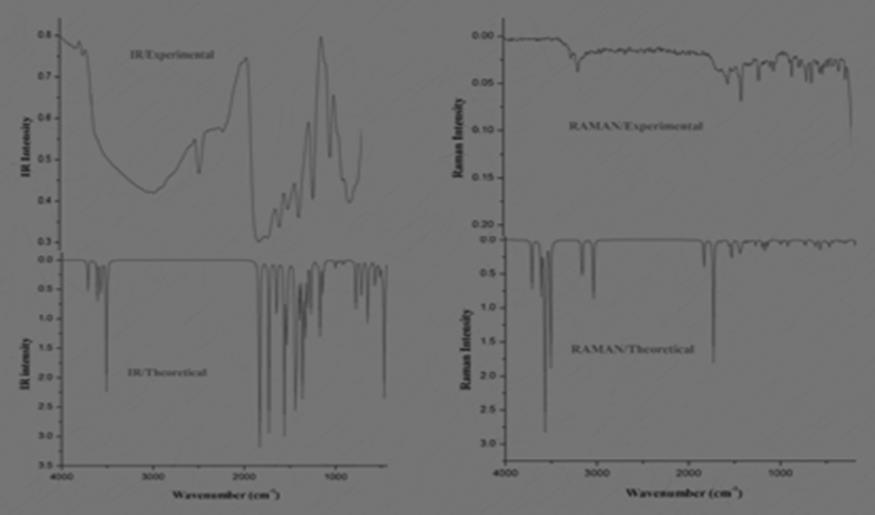
The correlation graph between the calculated and observed values is drawn by the RMS value obtained by using the below eq. (1) correlation coefficient of the study compound is determined and it is represented in Figure 3.
Figure 3.Correlation Graph of OTTHPYHCT between Experimental and Theoretical Value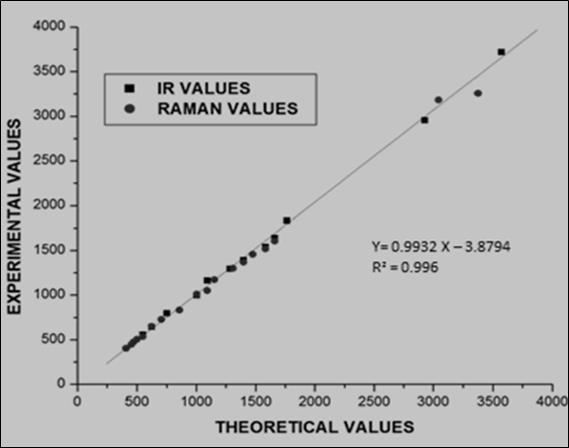
RMS= — 1
— 1
The RMS error of the observed IR and Raman bands are found to be 23.83, 12.05 for DFT method. The small differences in observed and calculated vibrational modes are due to the performance of calculation in the gaseous phase.
The proton NMR shows NH Hydrazide peak at 7.908 and NH RING peak at 10.218, 11.923. The active methylene hydrogen CH2 methylene is confirmed by the presence of a sharp peak at 4.64. The amine NH2 proton noted at the substituted chain gives a peak at the range 9.48 respectively.
13C-NMR spectrum gives peaks at 183.3, 175.39, 166.45, 154.81 and 82.49 for respective carbon atoms present in the functional group of the molecule. The visual presentation of 1HNMR and 13CNMR are shown in Figure 4 and Figure 5.
Figure 4.1H-NMR spectra of OTTHPYHCT
Figure 5.13C-NMR of OTTHPYHCT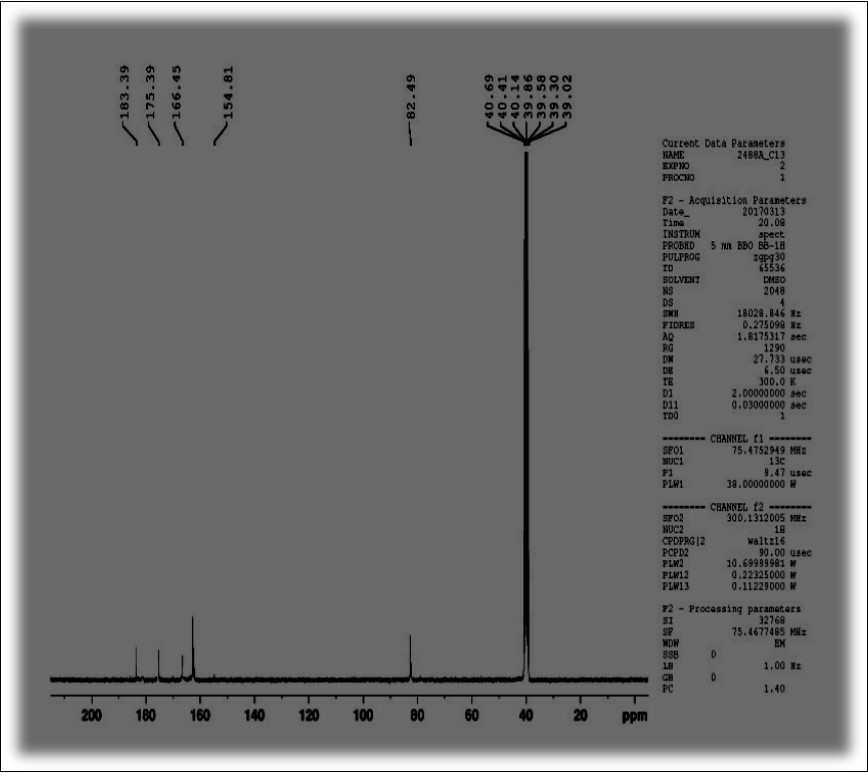
Molecular Geometry:
The optimized geometric parameters of OTTHPYHCT calculated by DFT/B3LYP/6-31G (d, p) are represented in Table S2 shown with atom numbering in Figure 6. The molecule under investigation consists of one ring and a substituent chain is attached. The ring and substituent chain are attached by C=N hydrazone moiety. The introduction of the new substituent in the ring results in distortion of the ring from the planar position and thereby the bond angle, bond length of the compound after substitution varies from the parent compound 12.
Figure 6.Optimized structure of OTTHPYHCT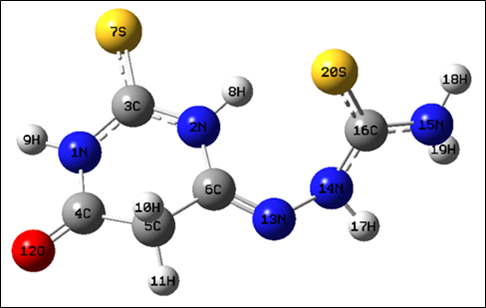
The bond length of C6-N13 of hydrazone linkage is about 1.2855 A whereas the C-N bonds values are nearly about 1.3 A . The reason may be hybridization where C=N is Sp2 hybridized and C-N SP3 hybridized. Lone pair atoms show the difference in bond length i.e. the bond length of C=O are shorter than C=S mainly due to polarizing nature. N-H and C-H bond length are calculated to be nearby 1 A . The dihedral angles of N2-C6-N13, C5-C6-N13, C6-N13-N14 are 127.2 A, 116.9 A , 118.8 A which is in agreement with the literature 13. All the bond length and bond angles values of functional groups present in the compound are tabulated in Table S2. A small difference is observed between computed value and experimental literature value as computed deals with gaseous phase and the other deals with the solid phase.
whereas the C-N bonds values are nearly about 1.3 A . The reason may be hybridization where C=N is Sp2 hybridized and C-N SP3 hybridized. Lone pair atoms show the difference in bond length i.e. the bond length of C=O are shorter than C=S mainly due to polarizing nature. N-H and C-H bond length are calculated to be nearby 1 A . The dihedral angles of N2-C6-N13, C5-C6-N13, C6-N13-N14 are 127.2 A, 116.9 A , 118.8 A which is in agreement with the literature 13. All the bond length and bond angles values of functional groups present in the compound are tabulated in Table S2. A small difference is observed between computed value and experimental literature value as computed deals with gaseous phase and the other deals with the solid phase.
Frontier Molecular Orbital and Global Reactivity Descriptors:
FMO analysis using computational methods is widely employed to explain the electronic as well as the optical properties of organic compounds 14. According to the frontier molecular orbital theory FMO, the shapes and symmetries of the highest occupied and lowest unoccupied molecular orbitals HOMO and LUMO are crucial in predicting the reactivity of a species and the stereo chemical and regiochemical outcome of the chemical reaction. The electronic reconfiguration and electronic excitations helps in studying the electric and optical properties of the organic molecules. Overlapping of molecular orbitals decides the stabilization of the bonding orbital and destabilization of the antibonding molecular orbital. Here, irrespective of HOMO and LUMO, the s and p orbital lobs were interlinked and the electron clouds were shared their spatial orbital’s and also spent their time in the place of link. During that time, the analogous physical and chemical properties mutually shared between the atoms. The charge levels were fluctuated due to the electron cloud sharing. A typical electron density distribution of HOMO and LUMO for studied compounds is shown in Figure 7. The HOMO of is OTTHPYHCT mainly located around the substituted thiosemicarbazide linkage, LUMO is located over the C=O, C=S and thiobarbituric ring. From the Figure 7, it could be seen that HOMO of OTTHPYHCT is mainly located over C=S moiety, LUMO of this molecule is in the C=O, C=S moiety and C=N bonded to ring. The energy gap obtained is about 4.1868 eV. And the HOMO-LUMO orbital values are represented in Table S3.
Figure 7.HOMO-LUMO diagram of OTTHPYHCT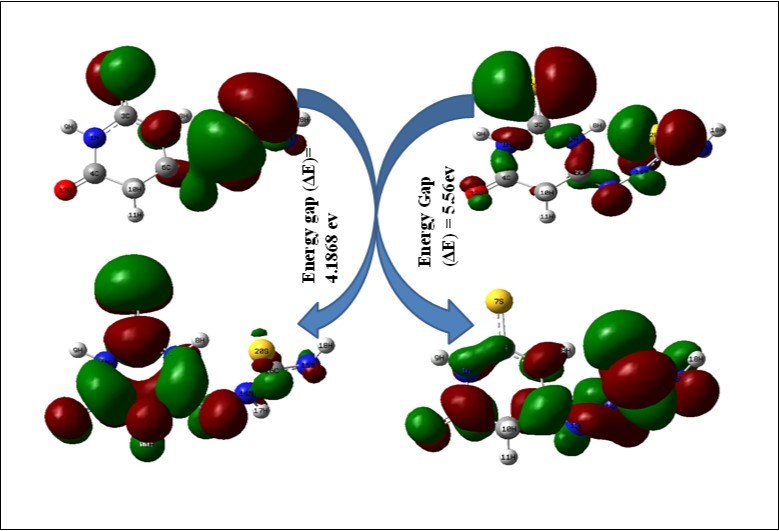
To compute the reactivity of the synthesized thiosemicarbazone OTTHPYHCT we also evaluate various global reactivity indices, like the chemical potential, chemical hardness, electrophilicity and nucleophilicity 15, 16, 17, 18, 19, 20, 21, 22.
The vertical ionization potentials IPs and electron affinities EAs were obtained from the HOMO-LUMO values, which can be used to determine the global electrophilicity value ω. It is a measure of the energy stabilization of the system. The global electrophilicity value was calculated as μ2/2η 23, where μ is the chemical potential approximated as - IP +EA/2, and η is the chemical hardness approximated as IP - EA. Chemical hardness measures the resistance to change in the electron distribution in a collection of nuclei and electrons 24. The calculated values of the chemical potential, chemical hardness and global electrophilicity are 4.12 eV, 2.09 eV and 4.05 eV, respectively. The value of η for thiosemicarbazone OTTHPYHCT is small, which indicates that it is relatively soft on the scale of hardness.
According to the definition, the electrophilicity index is a measure of the propensity of chemical species to accept electrons. The reactivity of the nucleophile is characterized by a lower value of μ, ω, and conversely a good electrophile is characterized by a high value of μ, ω. This new reactivity index measures the stabilization in energy when the system acquires an additional electronic charge ΔN max from the environment 25.
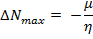
— 2
The maximum charge transfer ΔN max towards the electrophile was evaluated using Eq. (2). Thus, while the quantity defined by S= 1/ɳ describes the softness of the molecule i.e. Eq. (2) describes the charge capacity of the molecule. Very recently, Ayers and co-workers 26, 27 have proposed two new reactivity indices to quantify nucleophilic and electrophilic capabilities of a leaving group, nucleofugality ΔEn and electrofugality ΔEe, defined as follows,
 — 3
— 3

— 4
From the above mathematical formulation the electronic charge ΔN max, reactivity indicesnucleofugality ΔEn and electrofugality ΔEe are found to be 1.97, 9.22 and 0.98.
Natural Bond Analysis:
NBO analysis is an efficient tool for investigation of charge transfer in a molecular system 28. The lone pair atoms nitrogen, oxygen and sulphur present in the molecule results in hyper conjugative interaction which helps in stabilization of the molecule. From NBO Table S4 the energies of n-π* transition N1→C3-S7, N1→C4-O12, N2→C3-S7, N2→C6-N13, N14→C16-S20 are larger and are found to be 269.37, 207.32, 292.42, 174.64, 213.76 KJ/mol probably when compared with n-* transition which shows lower energy values for N15→C16-S20, O12→N1-C4, O12→C4-C5, N13→N2-C6, S7→N1-C3, S7→N2-C3, C5, S20→N14-C16, S20→N15-C16, C16→S20→C16-S20 donor to acceptor and the energy values are as follows 195.77, 121.5, 88.74, 68.16, 49.37, 48.37, 43.14, 31.71 KJ/mol. The above interaction in the molecule results in weakening of the respective bond and makes the entire molecule highly polarized which helps in NLO study of the considered molecule. Further from the above electron transition from donor to acceptor clearly indicates the flow of electron from parent thiobarbituric ring to the thiosemicarbazide substituent attached to the ring.
Hyperpolarizability, Non-Linear Optical Studies Nlo and Thermodynamic Properties:
Materials having high nonlinear optics NLO responses are very useful in optoelectronic. Devices and non-linear optics have great applications in information technology, and in other Industries. NLO materials also have wide range applications in photonic communication, digital memory devices, national defense and in pharmaceutical industry29.
Compounds having electron donor and electron acceptor groups along with a conjugated system can be considered as strong candidates for such applications. In order to investigate the relation between molecular structure and NLO properties, the first hyperpolarizibility value was simulated at the B3LYP/6-31G d, p level of theory along with the additional keyword POLAR and mathematically calculated using the following equation: The value of the first hyperpolarizibility is determined in a.u. and then converted to esu using the conversion factor 1 a.u. = 8.6393X10-33 esu. The calculated first hyperpolarizability parameters of the compound aregiven in Table S5. As reflected from the data, the first hyperpolarizability values show the same trend according to the electron donating and withdrawing capacity, and extended conjugation pattern.
On the basis of vibrational analysis at B3LYP/6-31G d, p level, the standard statistical thermodynamic functions: heat capacityC0 p, m, entropy S0 m and enthalpy changesH0 m for the title compound were obtained from the theoretical harmonic frequencies listed in Table S6. It can be observed that there thermodynamic functions are increasing with Temperature ranging from 100-1000K due to the fact that the molecular vibrational intensities increase with temperature 30 and it is graphically represented in the Figure 8.
Figure 8.Thermodynamic Plot of Otthpyhct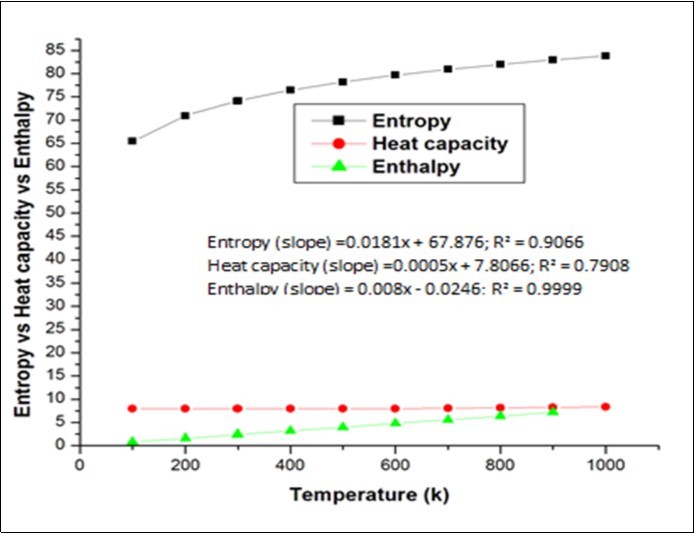
Conclusion:
The synthesized thiosemicarbazone compound 2 - 6 – oxo - 2-thioxotetrahydropyrimidin – 4 1H - ylidene hydrazine carbothioamide was confirmed by spectral data. UV spectra show n-π*, n-σ* transition due to lone pair atoms present in the compound. IR, Raman, 1HNMR and 13CNMR datas helps in conformation of formation of the molecule by C=N linkage assignment. The molecular geometry, bond parameters and vibrational assignment using DFT/ B3LYP/ 6-31G d, p method are briefly elucidated. From HOMO-LUMO the energy gap is determined and it helps in explaining the reactivity nature and stability of the compound. From the energy values of HOMO-LUMO the global reactivity descriptors η, µ, ω, electronic charge ΔN max, nucleofugality ΔEn, electrofugality ΔEe were calculated. Natural bond analysis explains the flow of electrons from ring to the substituent attached and the energy responsible for the stabilization of the molecule. The first hyperpolarizability value is β0 = 2.048*10-30 esu which is compared with the standard urea value and it is found to be 5 times greater. In addition to this thermodynamic details show a gradual increase with increase in temperature due to increase in vibrational intensities inside the molecule in gas phase.
References
- 2.Koch O, Stuttgen G. (1950) Naunyn-Schmiedebergs Archiv for Experimentelle Pathologie and Pharmacology. 210, 409-423.
- 6.N K Singh. (2001) . Proceedings of the Indian Academy of Sciences: Chemical Sciences 113, 257-273.
- 9.P J Stephens, F J Devlin, Chabalowski C F, Frisch M J. (1994) . , Journal of Physical Chemistry 98, 11623-11627.
- 15.Martínez-Araya J I, Salgado-Morán G, Glossman-Mitnik D. (2013) . , J. Phys. Chem. B 117, 6639-6651.
- 23.Parr R, Yang W. (1984) Density Functional Approach to the Frontier-Electron Theory of Chemical Reactivity. , J. American Chemical Soc 106, 4049-4050.
- 26.Ayers P W, Anderson JSM, Bartolotti L J. (2005) doi: 10.1002/qua.20307.Int.J.Quantum Chem. 101, 520.
Cited by (4)
This article has been cited by 4 scholarly works according to:
Citing Articles:
R. Akkaya, Birnur Akkaya, R. O. Bello - Journal of Molecular Structure (2022) Semantic Scholar
Journal of Molecular Structure (2022) OpenAlex
H. I. Umar, A. Ajayi, R. O. Bello, H. O. Alabere, A. A. Sanusi et al. - Chemické zvesti (2021) Semantic Scholar
Chemical Papers (2021) OpenAlex