
Abstract
A multistep synthesis of 2-chloro-9-(2′-deoxy-2′-fluoro-β-D-arabinofuranosyl) adenine (clofarabine) is described from methyl β-D-ribofuranoside. A new improved method for preparation of 1,2-diacetyl D-ribofuranose derivative was developed via acetolysis of tri-O-pivaloylated D-ribofuranoside and plausible mechanism of the reaction was proposed. Synthesis of 3′,5′-di-O-pivaloyl-2,6-dichloropurine β-D-riboside along with isomeric 2′,5′-di-O-pivaloyl nucleoside was carried out by stereoselective glycosylation reaction of 2,6-dichloropurine with peracylated D-ribofuranose followed by regioselective 2´-O-deacetylation of protected β-ribonucleoside with different bases. Mild C2′-β-fluorination of the purine 3′,5′-di-O-pivaloyl ribonucleoside with an excess of diethylaminosulfur trifluoride afforded protected 2,6-dichloropurine 2′-fluoro β-D-arabinoside as the key intermediate. Efficient route to clofarabine was also investigated using anion glycosylation of 2-chloroadenine potassium salt with the 1-α-bromide and potassium tert-butoxide in binary solvent mixture, chromatography for separation of a mixture of anomeric nucleosides (a β/α ratio of 3.0:1) and deacylation of benzoylated 2′-fluoro β-nucleoside. Novel N6-isopentyl clofarabine analogue was synthesized by a direct alkylation of the parent nucleoside
Author Contributions
Academic Editor: Loai Aljerf, Department of Life Sciences, Faculty of Dentistry, University of Damascu.
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2022 Grigorii Sivets, et al.
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Introduction
Cytotoxic agents relating to a class of modified nucleosides are widely used as effective therapeutics for treatment of cancer 1, 2, 3, 4. Nucleoside antimetabolites as mimics of natural purine or pyrimidine counterparts are capable of disrupting normal DNA synthesis after incorporation in DNA or by promoting apoptosis, exert their cytotoxic effects and different mechanisms of biological action 3. Pharmacological studies and clinical trials of synthetic purine nucleosides led to discovery of several analogues as antitumor drugs 2, 5. Among a series of known purine arabinonucleosides with anticancer activities (Figure 1), fludarabine and clofarabine have found applications as clinical drugs for the treatment of hematologic malignances. Clofarabine (2-chloro-2′-fluoro-2′-deoxyarabinofuranosyl adenine, 3), potent cytostatic agent with increased stability of the glycosidic bond, is currently used as the antileukemic drug in therapy of pediatric patients with refractory and relapsed lymphoblastic leukemia 6, 7.
Figure 1.Structures of anticancer fluorinated purine nucleosides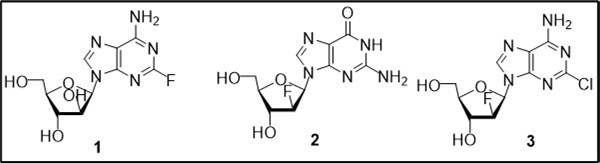
Multidirectional mechanism of cytotoxic action of clofarabine, that is not a substrate of adenosine deaminase, includes inhibition of synthesis of DNA and RNA, DNA polymerases, ribonucleotide reductase, and direct induction of cell apoptosis 7, 8. Clofarabine was shown to display in vitro activity against HIV-1 strain and possess dual inhibitory anti-HIV-1 mechanism in virus replication 9.
Various chemical and chemoenzymatic methods were explored for synthesis of anticancer nucleosides 2. The synthetic routes to purine C2′-β-fluorinated nucleosides and clofarabine associated with this class have been elaborated using different 2-deoxy-2-fluoro-D-arabinofuranose derivatives, acyclic C2-fluorinated dithioacetal precursors and selectively protected ribonucleosides 10, 11, 12, 13, 14. Bauta et al has developed practical and cost-effective synthesis of clofarabine via the stereoselective coupling reaction of 2-chloroadenine with the 1-bromosugar in the presence of potassium tert-butoxide followed by deprotection of the intermediate benzoylated β-nucleoside without exploiting chromatography 11. Cen and Sauve used silyl protected 2-deoxy-2-fluoro-D-arabinofuranose derivative, prepared by electrophilic fluorination of TIPS-protected 2-deoxyribonolactone, for efficient synthesis of clofarabine in six steps via coupling 2,6-dichloropurine with 1-α-chlorosugar 12. Chemoenzymatic approaches to synthesis of clofarabine were described by Mikhailopulo et al utilizing benzoylated 2-deoxy-2-fluoro-D-arabinofuranose and its 1-α-phosphate derivative 14. Stereospecific formation of N-β-glycosidic bond on the condensation step of the sugar phosphate with 2-chloroadenine and other purine bases catalysed by the recombinant E.coli nucleoside phosphorylase is a distinct feature of enzymatic methodologies studied 14, 15, 16 towards purine 2′-deoxy-2′-fluoro-β-D-arabinonucleosides compared to the known chemical methods 10, 11, 17. Another interesting synthetic route to purine 2′-deoxy-2′-fluoro-β-D-arabinonucleosides was developed by direct introduction of the C2′-β-fluorine function via nucleophilic fluorination of C2′-α-hydroxyl activated derivatives of 3′,5′-diprotected ribonucleosides with different fluorinating agents 18, 19, 20, 21.
As part of our efforts on synthesis of biologically active purine nucleosides from sugars, herein we report study of synthetic approaches to clofarabine using regio- and stereoselective transformations of pivaloylated D-ribose derivatives and nucleosides,N-glycosylation reactions of purine bases with available carbohydrate precursors, and novel purine modified analogues.
Results and Discussion
Synthesis of clofarabine was studied using two synthetic routes from known carbohydrate derivatives. In the first approach, we have employed peracylated D-ribofuranose 12 prepared by acetolysis reaction of methyl 2,3,5-O-pivaloylated β-D-ribofuranoside (5) taking into account our previous investigations in this field of 2́-β-fluoro-substituted adenine nucleosides 21 (Scheme 1). However, we failed to reproduce our synthetic procedure 21 reported for preparation of the diacetate 12 through acetolysis of acylated methyl β-D-riboside in a mixture of Ac2O/AcOH/H2SO4 (5.2% vol sulphuric acid) at room temperature. 1-O-Acetate 6 along with the target 1,2-diacetyl D-ribofuranose derivative 12 (21-45%), or only monoacetyl derivative, were prepared in a series of experiments (Scheme 1, conditions b). Further, we have undertaken study of acetolysis of 5 varying various reaction conditions in order to prepare 12 with high yield. Acetolysis (conditions c) in a mixture of reagents - AcOH, Ac2O, H2SO4 (8.0:1.0:1.1) with increased content of sulphuric acid (9.6% vol) gave rise to the 1-O-acetyl derivative 6 as a mixture of β/α-anomers (68%) and acyclic product 9 (10%). Mechanism of the formation of by-product 9 via protonation of the monoacetate 6 and generation of open furanose derivative 8 is outlined in Scheme 1. 1-O-acetate 6 was prepared from methyl ribofuranoside derivative 5 without formation of the diacetate 12, using anhydrous conditions for acetolysis reaction explored earlier for preparation of 1,2-di-O-acetyl-3,5-di-O-pivaloyl-L-arabinofuranoses from pivaloylated methyl L-arabinofuranoside 22. As result of studying acetolysis reaction we have found optimal conditions to obtain 12 with high yield (72%, conditions d) after chromatography on silica gel.
Scheme 1.Study of acetolysis reaction of pivaloylated methyl D-ribofuranoside 5 and proposed mechanism for the formation of the diacetate 12. Reagents and conditions: a) ref. 21, 89%; b) AcOH, Ac2O, H2SO4 (12.6:1.6 :1.0, vol), 5.2% H2SO4, rt, 3-4 h, 6, 20-41%; 12, 21-45%; c) AcOH, Ac2O, 9.6% H2SO4 (8.0:1.3:1.0, vol), 24 h, rt, 6, 68%; 9, 10%; d) AcOH, Ac2O, H2SO4 (8.05:1.0:1.1, ratio), 10.0% H2SO4 rt, 5 h, H2O, 90 min, 12, 72%;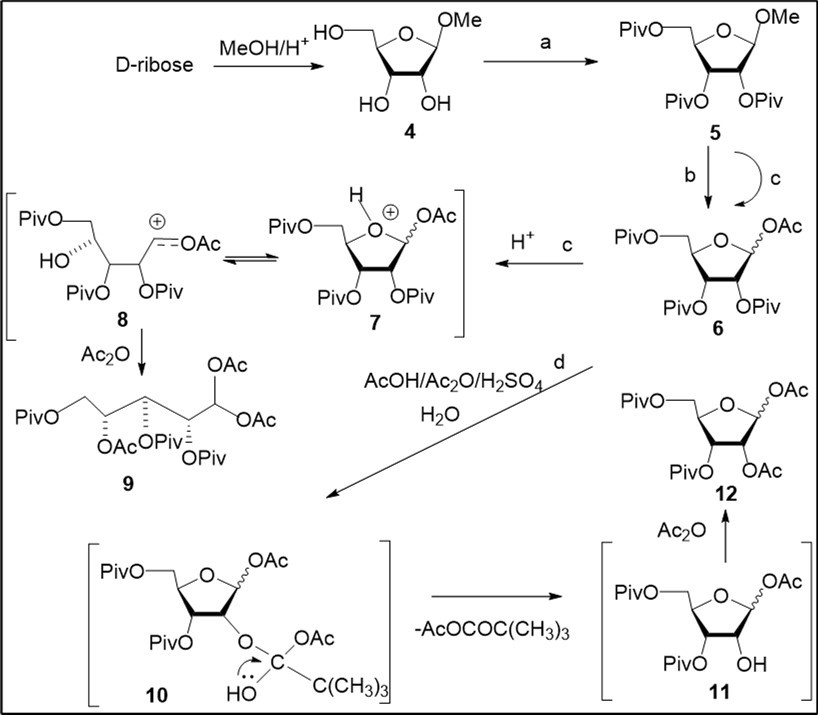
Carrying out acetolysis of 5 in the presence of a small quantity of water resulted to the target diacetate with a high yield. Proposed mechanism of the formation of 12 is shown in Scheme 1. We propose that synthetic route to the target acylated D-ribofuranose derivative includes formation of the 1-O-acetate 6 on the first step of acetolysis (Ac2O/AcOH/H2SO4) followed by generation, in the presence of water, intermediate D-ribofuranose derivatives 10 and 11, which lead to exchange of the 2-O-pivaloyl group in the starting riboside by the 2-O-acetyl group after acetylation at 2-hydroxyl group in selectively acylated ribofuranose 11 with acetic anhydride. It is important to note that the mechanism under above consideration for the acetolysis reaction of D-glycoside 5 with the formation of the 1,2-diacetate 12 differs from that suggested earlier in our previous work 22 for preparation of isomeric 1,2-di-O-acetyl-3,5-di-O-pivaloyl-L-arabinofuranoses via intermediate 1,2-pivaloyloxonium ion from protected L-arabinoside under anhydrous conditions.
Stereoselective glycosylation of silylated 2,6-dichloropurine with the 1,2-diacetate 12 in in acetonitrile in the presence of TMSOTf gave fully O-acylated β-D-riboside of 2,6-dichloropurine 13in 96% yield (Scheme 2).
Scheme 2.Synthesis of clofarabine 3 from the diacetate 12. Reagents and conditions: a) MeCN, silylated 2,6-diClPurine, TMSOTf, rt, 96%; b1-6) deacylation of 13, solvent/base, a mixture of nucleosides 14 and 15, 51-69% (Table 1); c) 14/15, DAST, CH2Cl2, Py, 00→rt, 59% for 16, 2-7% for 17; d) NH3/1,2-DME, rt, 18 h, 96%; e) MeONa/MeOH, rt, 74%.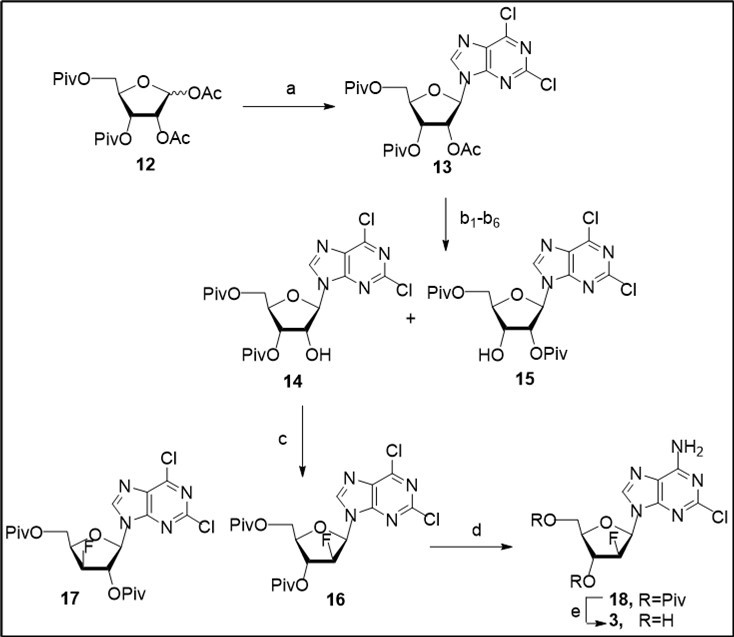
Selective removal of 2′-O-acetyl group in protected nucleoside 13 was explored under different conditions which are summarized in Table 1 (entries 1-6). Mild removal of 2′-O-acetyl group in nucleoside 5 with the known method, with an excess of NaHCO3 in methanol 10, gave rise to a mixture of 3′,5′-di-O-pivaloyl (14)and 2′,5′-di-O-pivaloyl (15) 2,6-dichloropurine β-D-ribosides, which were inseparable by column chromatography on silica gel, in an overall 51% yield. The deacetylation of 13 under the mild basic conditions was accompanied by migration the 3′-O-pivaloyl group of 14 from 3′-position to 2′-position with the formation of the 2′-O-pivaloylate 15 (a ratio of 14/15 – 1.7/2.0:1 according to 1H NMR data of the crude reaction mixtures). We failed to isolate individual acylated nucleosides 14 and 15 from a mixture using chromatography on silica gel or crystallization. Second known approach with application of dibutylthin oxide (Bu2SnO) in methanol 23, 24 was tested for selective removal of the 2́-O-acetyl group in nucleoside 13. Mixtures of 3′,5′-di-O-pivaloyl (14)and 2′,5′-di-O-pivaloyl (15) 2,6-dichloropurine ribosides were prepared in 55% and 69% yields, respectively, using 1.1 or 2.1 equiv of Bu2SnO in anhydrous methanol (entries 2-3), the best ratio of isomeric ribosides being achieved under refluxing of 13 with 2.1 equiv of Bu2SnO. Novel method for selective deacetylation of nucleoside 13 was explored using NaOCN under various conditions (solvent and excess of inorganic salt, entries 4-6). Best yield of a mixture of nucleosides 14/15 and regioselectivity of the deacylation reaction (a ratio of 14/15 –2.2:1) was prepared with 1.9 equiv of sodium cyanate in anhydrous methanol at room temperature (entry 5). The regioselective deacetylation of 13 with NaHCO3, NaCNO or Bu2SnO in methanol was accompanied by migration the 3′-O-pivaloyl group of 3′,5′-diprotected nucleoside 14 with the formation of the 2′-O-pivaloylate 15 (Table 1). Making use 4.3-fold excess of NaHCO3 or 1.9 eqiuv of NaCNO in anhydrous methanol, mixtures of 14/15 were prepared in 51% and 56% yields, respectively. The use of a large excess of NaHCO3 in methanol (entry 1) produced less yield of a mixture of isomeric nucleosides 14 and 15 than that isolated after selective deprotection of the 2′-O-acetate 13 with NaCNO (entry 5). These findings may be explained by different solubility of inorganic sodium salts at room temperature that is the important factor in development of efficient method for cleavage of 2′-O-acetyl group. The increase of the reaction time in control experiment with an excess of NaCNO in anhydrous methanol resulted in the formation of by-products (according to TLC data and 1H NMR data of the reaction mixture) and reducing overall yield of isomeric nucleosides. Fast removing of 2´-O-acetyl protecting group in the intermediate nucleoside 13 (entry 5) likely proceeds by a transesterification mechanism via generation of methoxide anion from sodium cyanate in anhydrous methanol with a gradual release of HOCN.
Table 1. Study of regioselective deacetylation of 2´-O-Ac-2´,3´-di-O-pivaloyl ribonucleoside of 2,6-dichloropurine 13 under various conditions.| Entry | Reaction conditions | Ratio of reagent: nucleoside 13 | Reaction time (h) | Ratios of isomeric nucleosides 14 and 15a | Rate of conversion of 2´-O-acetate 13 | Yieldb (%) |
| 1 | NaHCO3/MeOH | 4.3:1 | 1.8 h | 1.80:1.0 | 73 | 51 |
| 2 | Bu2SnO/MeOH | 1.1:1 | 7.2 h | 2.00:1.0 | 71 | 55 |
| 3 | Bu2SnO/MeOH | 2.1:1 | 4h50 min | 2.44:1.0 | 85 | 69 |
| 4 | NaOCN/MeOH | 1.2:1 | 2h20 min | 2.00:1.0 | 75 | 66c |
| 5 | NaOCN/MeOH | 1.9:1 | 42 min | 2.20:1.0 | 81 | 67 |
| 6 | NaOCN/MeOH/THF | 1.9:1 | 46 min | 2.20:1.0 | 79 | 60 |
Further, introduction of the C2′-β-fluorine atom via the nucleophilic replacement of an activated C2′-α-hydroxyl of 3′,5′-di-O-pivaloyl ribonucleoside 14 containing bulky directing groups with a fluoride anion was studied on the next step. The treatment of a mixture of selectively blocked ribonucleosides 14 and 15 (a ratio - 2.3:1 after column chromatography on silica gel)with an excess of DAST in CH2Cl2/pyridine at room temperature afforded protected 2′-β-fluoro nucleoside(16,59%).Isomeric 3′-β-fluoro(17, 2-7%)nucleoside was also isolated as by product under the fluorination step of mixtures of nucleosides 14 and 15 after column chromatography on silica gel. New 2,6-dichloropurine 3′,5′-di-O-pivaloyl β-D-2′-fluoroarabinonucleoside 16 was prepared as the key intermediate for synthesis of different purine modified 2′-β-F-nucleosides.Analysis of conformational peculiarities of selectively protected 2,6-dichloropurine ribonucleoside 14 based upon its 1H NMR spectral data (J1',2´ = 6.25, J2´,3´ =5.6, J3´,4´ =3.27 Hz) shows that the pentofuranose ring in this nucleoside takes a C-2′-endo conformation, S-type, as in the case of the adenine analogue 21 studied earlier in the DAST reaction which leads to adenine 2′-β-fluoro-nucleoside under reflux in moderate yield. However, despite the population of S-type conformation in N/S pseudorotational equilibrium is predominant in solution for the both 3′,5′-di-O-acyl purine ribonucleosides, there are some differences in the spatial organization of 3′,5′-di-O-pivaloyl-2,6-dichloropurine nucleoside and the corresponding adenosine derivative, their C2′-α-hydroxyl activated intermediates forming with DAST during the course of fluorination reactions. Selective amination of 2,6-dichloropurine derivative 16 with ammonia in 1,2-dimethoxyethanefollowed by the removal of acyl groups in 3′,5′-di-O-pivaloyl 2′-β-fluoro nucleoside of 2-chloroadenine 18 with sodium methoxide in methanol resulted in the target nucleoside 3 in 71% combined yield on two steps after column chromatography on silica gel. Thus, synthesis of clofarabine was carried out in seven steps from D-ribose in moderate overall yield exploiting the stereoselective glycosylation of 2,6-dichloropurine with available peracylated D-ribose, selective 2′-O-deacetylation of the intermediate tri-O-acylated nucleoside, and the DAST fluorination of 3′,5′-di-O-pivaloyl-2,6-dichloropurine ribonucleoside.
Next, a short approach to clofarabine (Scheme 3) was investigated from the glycosyl bromide 19 prepared by mild bromination of available perbenzoylated 2-deoxy-2-fluoro-α-D-arabinofuranose with TMSBr 17 taking into account the cost-effective synthesis reported earlier 11. Anion glycosylation of 2-chloroadenine salt, generated in the presence of potassium tert-butoxide and anhydrous potassium bromide as additive in 1,2-dimethoxyethane, with the 1-α-bromide 19 in acetonitrile resulted in a mixture of N9-β-anomer 20 (48%) and its α-anomer 21 (16%) which were separated by column chromatography on silica gel. Formation of protected β- and α-nucleosides (β/α ratio - 3:1) along with by-product (7-8%) was observed in this experiment according to 1H NMR data of the crude reaction mixture in CDCl3 (Scheme 2, conditions a1).
Scheme 3.Synthesis of clofarabine 3 and its N6-alkylated analogue 23 from the glycosyl bromide 19. Reagents and conditions: a1) bromide 19, K-salt of 2-ClAde generated with t-BuOK in 1,2-DME in the presence of 1.6 equiv anhydrous KBr, CH3CN, rt, 18 h, 45-48% 20, 16%, 21; a2) bromide 19, K-salt of 2-ClAde generated with t-BuOK in 1,2-DME in the presence of 1.5 equiv anhydrous KBr, CH3CN/CH2Cl2 (2:1), rt, 55 h, 55% 20, 18% 21; b) 20, NH3/MeOH, rt, 78%; c) 3, t-BuOK, DMSO, (CH3)2CHCH2CH2OMs (22), 89-90 0C, 90 min, 23, 73% taking into account of recovery of the starting nucleoside.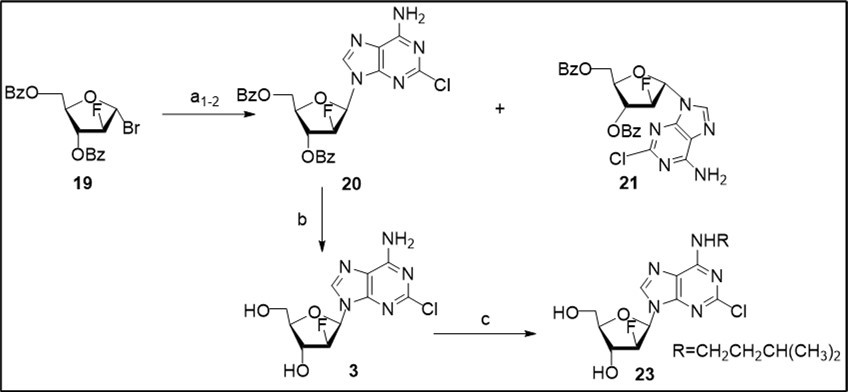
The coupling of 19 with 2-chloroadenine salt, produced by treatment with t-BuOK in the presence of potassium bromide, in a mixture of CH3CN/CH2Cl2 (2:1) (Scheme 2, a2) at room temperaturegave rise to β-(55%) and α-(18%) protected nucleosides after column chromatography on silica gel with a similar β/α ratio and higher overall yield in comparison to the previous reaction conditions. No increase of anomeric stereoselectivity of the glycosylation reaction of 2-chloroadenine salt was observed in a mixture of CH3CN/CH2Cl2 (1:1) in the presence of calcium hydride as additive and conversion of bromide was 86% for 36 h in this experiment. Synthesis of anomeric benzoylated nucleosides by anion glycosylation of 2-chloroadenine with 1-α-bromide involves competitive SN2 and SN1-reaction pathways, the latter occurs via generation of an oxonium ion intermediate. Formation of α-glycosylated product 21 in CH3CN/CH2Cl2 with potassium bromide as additive likely to proceed via a 3,5-benzoxonium ion 25 deriving from the 1-α-bromide, and a subsequent nucleophilic α-attack of the nucleobase at C1.
Characteristic of the studied glycosylation reactions of the potassium salt of 2-chloroadenine, generated using t-BuOK in 1,2-dimethoxyethane, with 1-α-bromosugar are: i) the anion glycosylation reaction with a small excess of the heterobase proceeds in heterogeneous phase in acetonitrile or a mixture of solvents with different polarities at room temperature with a full conversion of bromosugar to nucleoside products; ii) KBr as additive results in a little increase of β/α-anomeric selectivity towards protected N9-β-nucleoside, and iii) good isolated yield of intermediate benzoylated β-2′F-arabinonucleoside is achieved after selective glycosylation in a binary solvent system and separation of N9-β- and α-glycosylated products by column chromatography on silica gel in detected conditions. Furthermore, based on our previous results on efficient synthesis of benzoylated 2-amino-6-chloropurine 2′-fluoro-β-arabinonucleoside from the bromide 1917 and the above findings tested for isomeric 2-chloroadenine nucleoside 20, we may conclude that a better solubility of the modified purine base in organic solvents provide a higher β/α-stereoselectivity of heterogeneous glycosylation reaction of the purine potassium salt in a binary solvent mixture.
Deprotection of benzoylated 2′-fluoro β-nucleoside 20 with ammonia in methanol yielded clofarabine 3 in 78% yield after crystallization. The target nucleoside was prepared in 35-43% overall yields using column chromatography. Synthesis of N6-substituted clofarabine derivative 14 was carried out by a direct alkylation of 3 with 1-methanesulfonyloxy-3-methylbutane (22) in anhydrous dimethylsulfoxide in the presence of t-BuOK at 89-900C. The use of bulky alkylating agent prepared by mesylation of isoamyl alcohol with mesyl chloride resulted in a selective reaction on an exocyclic N6-amino group of the starting nucleoside under studied conditions to give new N6-isopentyl clofarabine analogue 23 in 73% yield after column chromatography on silica gel.
Conclusion
In summary, multistep synthesis of clofarabine and C2′-β-fluorinated purine modified nucleosides was accomplished using regio- and stereoselective reactions of O-pivaloylated D-ribose derivatives and mild fluorination of 3′,5′-di-O-pivaloyl-2,6-dichloropurine riboside containing bulky directing groups with DAST on the key step. Optimized method for preparation of the 1,2-diacetyl D-ribofuranose derivative was developed via acetolysis of tri-O-pivaloylated D-ribofuranose. The intermediate selectively protected purine nucleoside was obtained by stereoselective glycosylation reaction of the 1,2-diacetate with 2,6-dichloropurine followed by regioselective 2´-O-deacetylation of fully protected β-ribonucleoside with different deacylating reagents. The efficient two-step synthetic route to clofarabine via selective anion glycosylation of 2-chloroadenine with 3,5-di-O-benzoyl-2-deoxy-2-fluoro-α-D-arabinofuranosyl bromide in a mixture of solvents is described under mild reaction conditions. N6-isopentyl clofarabine derivative with potential biological activity was prepared by a direct alkylation of the parent nucleoside with 1-methanesulfonyloxy-3-methylbutane in the presence of t-BuOK.
Material and Methods
Column chromatography was performed on silica gel 60 H (70-230 mesh; Merck, Darmstadt, Germany), and thin-layer chromatography (TLC) on Merck silica gel aluminum 60 F254 precoated plates. The anhydrous solvents were distilled over CaH2, P2O5 or magnesium prior to the use. All commercially available reagents were used without further purification. 1H, 13C, and 19F NMR spectra were recorded in CDCl3, CD3OD and DMSO-d6 with a Bruker Avance-500-DRX spectrometer at 500.13, 126.76 and 470.59 MHz, respectively. 1H and13C NMR chemical shifts (δ, ppm) are relative to internal chloroform peak (7.26 ppm for 1H and 77.0 for 13C NMR). Chemical shifts are also reported downfield from internal SiMe4 (1H) or external CFCl3 (19F). Splitting patterns were reported as following: s: singlet, d: doublet, t: triplet, m: multiplet. J values are reported in Hz. Melting points were determined on a Boetius apparatus and were uncorrected. High resolution mass spectra (HRMS) were recorded on an Agilent Q-TOF 6550 Instrument (USA) using ESI (electrospray ionization).
Experimental Procedures
Synthesis of 1-O-acetyl-2,3,5-tri-O-pivaloyl-D-ribofuranose (6) and 1,1,4-tri-O-acetyl-2,3,5-tri-O-pivaloyl-D-ribose hydrate (9)
Methyl D-ribofuranoside 5 (0.6 g, 1.44 mmol) was coevaporated with anhydrous toluene (2x20 ml), dissolved in a mixture of CH3COOH (4.35 ml), Ac2O (0.52 ml) and H2SO4 (0.29 ml), prepared previously by adding H2SO4 to mixture of CH3COOH and Ac2O and stirring the solution for 7 min. The reaction mixture was stirred at ambient temperature for 4 h. Then Ac2O (0.2 ml) was added to it and the reaction mixture was stirred for 60 min at room temperature and H2SO4 (0.25 ml) was added to prepared solution. After stirring at rt for 24 h prepared solution was diluted with ethyl acetate (45 ml), washed with cooled water (4x25 ml) and then dried over anhydrous Na2SO4, evaporated to dryness. The residue was chromatographed on a silica gel, using mixtures of 6:1, 5: 1 and 3:1 petroleum ether-EtOAc to afford a mixture of 1-O-acetyl derivatives 6 (0.436 g, 68%,α/β =1.2:1) as a syrup and acyclic product 9 (0.07 g 10%) as a syrup.
Compound 6, β-anomer. 1H NMR (CDCI3): δ 6.11 (br.s, lH, J1,2<1.0 Hz, H-1), 5.34 (dd, lH, H-2), 5.25 (dd, 1H, J3,4=1.6, H-3), 4.39-4.45 (m, lH, H-4), 4.26 (dd, lH, J5,5' =12.2, J5,4=3.2, H-5), 4.23 (dd, lH, J5',4 = 4.5, H-5'), for α- and β-anomers: 2.09 and 2.08 (2s, CH3CO), 1.24, 1.23, 1.22, 1.20 and 1.19 [5s, (CH3)3-CO].
Compound 6, α-anomer. 1H NMR (CDCI3): δ 6.46 (d, lH, J1,2 =3.8 Hz, H-l), 5.24 (dd, lH, H-2), 5.35 (dd, lH, 4.1, H-3), 4.32-4.38 (m, lH, H-4), 4.29 (dd, lH, J5,5' = 12.4, J5,4 = 4.6, H-5), 4.18 (dd, lH, J5',4 = 3.5, H-5'), for α- and β-anomers: 2.09 and 2.08 (2s, CH3CO), 1.24, 1.23, 1.22, 1.20 and 1.19 [5s, (CH3)3-CO].13C NMR (CDCl3): δ 178.08, 177.93, 177.54, 177.20, 176.87, 176.80 [(C=O, 6хСОС(СН3)3], 169.56 and 169.19 (C=O, СОСН3), 98.16, 79.60, 74.07, 70.11, 63.07 (C-1, C-2, C-3, C-4, C-5, alpha-anomer), 94.23, 83.01, 70.57, 70.22, 63.49 (C-1, C-2, C-3, C-4, C-5, beta-anomer), 38.86 [2xСОС(СН3)3], 27.25, 27.18, 27.15 and 27.09 [4xСОС(СН3)3], 21.08 and 20.99 (2xСОСН3). HRMS (EI): m/z calcd for C22H36O9(M+Na)+: 467.2252, found 467.2254.
Acyclic product 9, 1H NMR (CDCl3): δ 7.08 (d, lH, J1,2 =5.7 Hz, H-l), 5.43-5.49 (m, 2H, H-2 and H-3), 5.34-5.39 (m, lH, H-4), 4.42 (dd, lH, J5,5' = 11.6, J5,4 = 2.3, H-5), 4.10 (dd, lH, J5',4 = 6.3, H-5'), 2.16, 2.14, and 2.09 (3s, CH3CO), 1.26, 1.25 and 1.22 (3s, 3x3Н, 3хСОС(СН3)3]. 13C NMR (CDCl3): δ 177.86, 176.81 and 176.58 [(C=O, 3хСОС(СН3)3], 169.72 and 168.22 (C=O, 2xСОСН3), 85.82, 69.58, 69.35, 68.07, 61.58 (C-1, C-2, C-3, C-4, C-5),38.04, 38.97 and 38.83 [3xСОС(СН3)3], 27.05, 27.03 and 26.98 [3xСОС(СН3)3], 20.74 and 20.59 (2xСОСН3). HRMS (EI): m/z calcd for C26H42O12(M+Na)+: 569.2569, found 569.2574.
Synthesis of 1,2-di-O-acetyl-3,5-di-O-pivaloyl-α,β-D-ribofuranose (12)
Methy-2,3,5-tri-O-pivaloyl-β-D-ribofuranose5(0.387 g, 0.929 mmol) was coevaporated with anhydrous toluene (2x20 ml), dissolved in a mixture of CH3COOH (3.7 ml), Ac2O (0.32 ml) and H2SO4 (0.35 ml), prepared previously by adding H2SO4 to mixture of CH3COOH and Ac2O and stirring the solution for 7 min. The reaction mixture was stirred for 3 h at ambient temperature and for 2 h at 30 0C. Then H2O (0.043 ml) was added and the reaction mixture was stirred for 90 min at room temperature and diluted with ethyl acetate (45 ml), the prepared organic phase was washed with cooled water (4x40 ml) and then dried over anhydrous Na2SO4, and evaporated to dryness. The residue was chromatographed on a silica gel, using a mixture of 8:1, 6:1 and 5:1 petroleum ether-EtOAc to afford the diacetate 12 (0.27 g, 72%, β/α=1.3:1) as a syrup.
1,2-diacetate 12,β-anomer. 1H NMR (CDCI3): δ 6.17 (d, lH, J1,2 =1.4, H-1), 5.40 (dd, lH, J2,3= 4.9, H-2), 5.31 (dd, H, J3,4= 1.3 Hz, H-3), 4.36-4.40 (m, lH, H-4), 4.24 (dd, J5,5' = 12.1, J5,4=3.5, H-5), 4.27 (dd, 1H, J5′,4=4.7, H-5'), for α- and β-anomers: 2.154, 2.15, 2.14, and 2.09 (4s, CH3CO), 1.29, 1.28, 1.27, and 1.24 [4s, (CH3)3-CO].
1,2-diacetate 12,α-anomer.1H NMR (CDCI3): δ 6.43 (d, lH, J1,2 =4.4, H-1), 5.30 (dd, lH, J2,3= 6.3, H-2), 5.30 (dd, lH, J3,4=1.7, H-3), 4.41-4.49 (m, lH, J4,5= 3.1, H-4), 4.24 (dd, lH, J5,5'=12.2, J5,4=3.0, H-5), 4.19 (dd, 1H, J5',4=3.0, H-5').
13C NMR (CDCl3): δ 178.06, 177.92, 177.50, 177.13 [(C=O, 4хСОС(СН3)3], 169.74 and 169.26 (C=O, СОСН3), 98.37, 80.15, 74.27, 70.37, 63.43 (C-1, C-2, C-3, C-4, C-5, beta-anomer), 94.12, 82.43, 70.53, 69.69, 63.43 (C-1, C-2, C-3, C-4, C-5, alpha-anomer), 38.86 [2xСОС(СН3)3], 27.21, 27.17, 27.02 and 27.00 [4xСОС(СН3)3], 21.09, 21.05, 20.55 and 20.29 (4xСОСН3). HRMS (EI): m/z calcd for C19H30O9(M+Na)+: 425.1782, found 425.1780.
Synthesis of 2,6-dichloro-9-(2’-O-acetyl-3’,5’-di-O-pivaloyl-β-D-ribofuranosyl)-9H-purine (13)
To a mixture of the diacetate 12 (0.35g, 0.87 mmol) and silylated derivative of 2,6-dichloropurine, prepared by conventional procedure from 0.164 g (0.87 mmol) of 2,6-dichloropurine, in anhydrous CH3CN (5 ml), TMSOTf (0.2 ml, 1.2 mmol) was added and then the reaction mixture was stirred for 150 min at room temperature and poured into 5% aq NaHCO3. The mixture was extracted with CHCl3 (3x50 ml), combined extracts were washed with water, dried and evaporated. The residue was chromatographed on silica gel, eluting with mixtures of 3:1, 2:1 and 1:1 hexane-EtOAc to afford the nucleoside 13 (0.44 g, 96%) as syrup. 1H NMR (CDCl3): δ 8.29 (s, 1H, H-8), 6.20 (d, 1H, J1′,2′ = 6.2 Hz, H-1′), 5.76 (t, 1H,H-2′), 5.52 (dd, 1H, H-3′), 4.44-4.47 (m, 1H, H-4′). 4.43 (dd, 1H, H-5′), 4.35 (dd, 1H, H-5′′), 2.06 (s, 3H, CH3CO), 1.28 and 1.24 (2s, 2x9Н, 2хСОС(СН3)3]. 13C NMR (CDCl3): δ 178.0 and 177.2 [(C=O, 2хСОС(СН3)3], 169.37 (C=O, СОСН3), 153.6, 152.7, 152.5, 143.7, 131.5 (C-2, C-6, C-4, C-8, C-5), 86.4 (C-1′), 81.5 (C-3′), 73.5 (C-4′), 70.4 (C-2′), 63.3 (C-5′), 39.07 and 39.01 [2xСОС(СН3)3], 27.35 and 27.2 [2xСОС(СН3)3], 20.4 (СОСН3). HRMS (EI): m/z calcd for C22H28Cl2N4O7 (M+Na)+: 553.1233, found 553.1228.
Synthesis of 2,6-dichloro-9-(3’,5’-di-O-pivaloyl-β-D-ribofuranosyl)-9H-purine (14) and 2,6-dichloro-9-(2’,5’-di-O-pivaloyl-β-D-ribofuranosyl)-9H-purine (15)
Method 1
To solution of protected nucleoside 13 (0.233g, 0.438 mmol)in anhydrous methanol (12 mL) was added solid anhydrous NaHCO3 (0.16 g, 1.9 mmol) and prepared solution was stirring for 110 min at room temperature, then reaction mixture was neutralized with acetic acid and evaporated, coevaporated with anhydrous benzol (2 x 10 ml). The residue was chromatographed on silica gel, using a mixture of 3:1, 2:1 and 1:1 hexane-EtOAc to afford the starting nucleoside 0.062 g (27%) and a mixture of nucleosides 14 and 15 (0.11 g, 51%) as a syrup.
1H NMR of a mixture of 14 and 15 (CDCl3): δ 8.31 (s, 1H, H-8, compound 14), 8.27 (s, 0.37H, H-8, compound 15), 6.16 (d, 0.37H, J1′,2′ = 4.2 Hz, H-1′), 6.00 (d, 1H, J1′,2′ = 6.25 Hz, H-1′), 5.55 (dd, 0.37H,H-2′), 5.32 (dd, 1H, J3′,4′ = 3.27 Hz, H-3′), 4.97 (m, 1H, J2′,3′ = 5.6 Hz, H-2′), 4.72 (m, 0.37H, H-3′), 4.48-4.51 (m, 1H, H-4′), 4.37 - 4.67 (m, 2H-5′, H-5′ and H-5′′, H-4′), 1.31, 1.26, 1.21 and 1.20 (4s, 24.6Н, 4хСОС(СН3)3]. 13C NMR (CDCl3): δ 178.3, 177.96, 177.92 and 177.68 [(C=O, 4хСОС(СН3)3], 154.9, 152.5, 152.4, 152.3, 144.1, 131.4; 89.2, 83.5, 73.8, 72.2 (C-1′, C-3′, C-4′, C-2′), 87.3, 82.5, 75.7, 70.1 (C-1′, C-3′, C-4′, C-2′), 63.11 (C-5′), 39.1 and 38.9 [4xСОС(СН3)3], 27.21 and 27.14 [4xСОС(СН3)3]. HRMS (EI): m/z calcd for C20H26Cl2N4O6(M+Na)+: 511.1127, found 511.1227.
Method 2
To solution of protected nucleoside 13 (0.02g, 0.038 mmol)in anhydrous methanol (1.2 mL) was added Bu2SnO (0.008 g, 0.0402 mmol) and prepared solution was stirred under refluxing for 7h 15 min, then reaction mixture was evaporated. The residue was dissolved with EtOAc(35 ml), the organic layer was washed with water (2x10 ml) and dried over anhydrous Na2SO4, and then evaporated to dryness. The residue was chromatographed on silica gel, eluting with mixtures of 3:1, 2:1 and 1:1 hexane-EtOAc to afford a mixture of nucleosides 14 and 15 (0.010 g,55%) as a syrup.
Method 3
To solution of protected nucleoside 13 (0.022g, 0.041 mmol)in anhydrous methanol (2 mL) was added Bu2SnO (0.022 g, 0.0884 mmol) and prepared solution was stirred under refluxing for 4h 50 min, then reaction mixture was evaporated. The residue was dissolved with EtOAc(30 ml), the organic layer was washed with water (2x10 ml) and dried over anhydrous Na2SO4, and then evaporated to dryness. The residue was chromatographed on silica gel, eluting with mixtures of 4:1, 3:1 and 2.5:1 petroleum ether-EtOAc to afford the starting nucleoside (0.003 g, 14 %) and a mixture of nucleosides 14 and 15 (0.012 g, 69%) as a syrup with recovery of the starting nucleoside.
Method 4
To solution of protected nucleoside 13 (0.021 g, 0.039 mmol) in anhydrous methanol (4 mL) was added NaOCN (0.003 g, 0.046 mmol) and prepared solution was stirred under for 140 min at room temperature. Then reaction mixture was diluted with EtOAc(30 ml), the organic layer was washed with water (2x10 ml) and dried over anhydrous Na2SO4, evaporated to dryness. The residue (0.02 g) contained the starting nucleoside (25%) and a mixture of nucleosides 14 and 15 (66%) according to 1H NMR data of the reaction mixture measured in CDCl3.
Method 5
To solution of protected nucleoside 13 (0.052 g, 0.0978 mmol) in anhydrous methanol (3 mL) was added NaOCN (0.006 g, 0.092 mmol) and prepared solution was stirred under for 22 min at room temperature, then sodium salt (0.006 g, 0.092 mmol) was added to prepared solution. The reaction mixture was stirred for 20 min and acetic acid (0.1 ml) was added, then evaporated, coevaporated with anhydrous toluene. The residue was dissolved with EtOAc(30 ml), the organic layer was washed with water (10 ml) and dried over anhydrous Na2SO4, and evaporated to dryness. The residue was chromatographed on silica gel, eluting with mixtures of 3:1, 2:1 and 1:1 hexane-EtOAc to afford the starting nucleoside (0.009 g, 17%) and a mixture of nucleosides 14 and 15 (0.027 g, 67%) as a syrup with recovery of the starting nucleoside.
Method 6
To solution of protected nucleoside 13 (0.043g, 0.081 mmol) in anhydrous methanol 2.9 mL) was added NaOCN (0.01 g, 0.154 mmol) and prepared solution was stirred under for 30 min at room temperature, then anhydrous THF (1.8 ml) was added. The reaction mixture was stirred for 15 min and acetic acid (0.1 ml) was added, then evaporated, coevaporated with anhydrous toluene. The residue was chromatographed on silica gel, eluting with mixtures of 3:1, 2:1 and 1:1 hexane-EtOAc to afford the starting nucleoside (0.008 g, 19%), a mixture of nucleosides 14 and 15 (0.019 g, 60%) as a syrup with recovery of the starting nucleoside.
Synthesis of 2,6-dichloro-9-(3¢,5¢-di-O-pivaloyl-2¢-deoxy-2¢-fluoro-β-D-arabinofuranosyl)-9H-purine (16) and 2,6-dichloro-9-(2¢,5¢-di-O-pivaloyl-3¢-deoxy-3¢-fluoro-β-D-xylofuranosyl)-9H-purine (17)
To a solution of a mixture of nucleosides 14 and 15 (0.12 g, 0.245 mmol, a ratio - 2.3:1) in anhydrous CH2Cl2 (4.5 ml) and pyridine (0.06 ml) was added 0.16 ml (1.22 mmоl) DAST at 0 0C. The reaction mixture was stirred for 60 min under cooling and then for 18 h at 25-300C. The residue prepared after treatment of reaction mixture was chromatographed on silica gel, using a mixture of 8:1, 5:1 and 3:1 hexane-EtOAc to afford protected 2´-β-fluoro nucleoside 16 (0.049 g, 59%) as a syrup and 3´-β-fluoro nucleoside 17 (0.003 g, 7%) as a syrup.
2´-β-fluoro nucleoside 16. 1H NMR (CDCl3): δ 8.37 (d, 1H, JH-8, F-2′ = 2.9 Hz, H-8), 6.48 (dd, 1H, J1′,2′ = 3.8 Hz, J1′,F-2′ = 21.7 Hz H-1′), 5.37 (dd, 1H, J3′,2′ = 2.7 Hz, J3′,F-2′ = 17.6 Hz, H-3′), 5.19 (dd, 1H, J2′,F-2′ = 50.3 Hz, H-2′), 4.53 (dd, 1H, H-5′), 4.41 (dd, 1H, H-5′′), 4.23-4.27 (m, 1H, H-4′). 1.29 and 1.24 (2s, 2x9Н, 2хСОС(СН3)3]. 13C NMR (CDCl3): δ 178.2 and 177.2 [(C=O, 2хСОС(СН3)3], 153.4, 152.5, 152.3, 130.7 (C-2, C-6, C-4, C-5), 145.2 (d, JC-8,F-2′ = 5.9 Hz, C-8), 92.7 (d, JC-2′,F-2′ =193.4 Hz, C-2′), 83.9 (d, JC-1′,F-2′ =16.9 Hz, C-1′), 81.8 (C-4′), 75.9 (d, JC-3′,F-2′=20.3 Hz, C-3′), 62.6 (C-5′), 39.0 and 38.9 [2xСОС(СН3)3], 27.27 and 27.1 [2xСОС(СН3)3]. 19F NMR (CDCl3): δ -197.5 (dt, F-2′). HRMS (EI): m/z calcd for C20H25Cl2FN4O5 (M+Na)+: 513.1084, found 553.1064.
3´-β-fluoro nucleoside 17.1H NMR (CDCl3): δ 8.36 (s, 1H, H-8), 6.26 (d, 1H, J1′,2′ = 1.9 Hz, H-1′), 5.52 (dd, 1H, J2′,F-3′ = 15.2 Hz, H-2′), 5.22 (dd, 1H, J3′,4′ = 1.9 Hz, J3′,F-3′ = 49.3 Hz, H-3′), 4.43-4.54 (m, 3H, H-4′, H-5′ and H-5′′), 1.32 and 1.28 (2s, 2x9Н, 2хСОС(СН3)3]. 13C NMR (CDCl3): δ 178.0 and 176.6 [(C=O, 2хСОС(СН3)3], 153.5, 152.6, 152.2, 143.4, 143.3, 130.8 (C-2, C-6, C-4, C-5), 93.2 (d, JC-3′,F-3′ = 183.5 Hz, C-3′), 87.6 (C-1′), 80.4 (d, JC-4′,F-3′ = 20.9 Hz, C-4′), 79.7 (d, JC-2′,F-3′ = 31.9 Hz, C-2′), 60.53 (d, JC-5′,F-3′ = 10.0 Hz C-5′), 38.85 and 38.81 [2xСОС(СН3)3], 27.1 and 26.9 [2xСОС(СН3)3]. 19F NMR (CDCl3): δ -200.9 (ddd, F-3′). HRMS (EI): m/z calcd for C20H25Cl2FN4O5 (M+Na)+: 513.1084, found 553.1063.
Synthesis of 6-amino-2-chloro-9-(3¢,5¢-di-O-pivaloyl-2¢-deoxy-2¢-fluoro-β-D-arabinofuranosyl)-9H-purine (18)
A solution of nucleoside 16 (0.05 g, 0.1 mmol)in 1,2-dimethoxyethane (4 mL) saturated at 0oC with ammonia was kept for 18 h at room temperature and then evaporated. The residue was chromatographed on silica gel, eluting with CHCl3,then CHCl3:MeOH - 30:1 to afford nucleoside 18 (0.046 g, 96%) as an amorphous powder.1H NMR (CDCl3): δ 8.03 (d, 1H, JH-8, F-2′ = 3.1 Hz, H-8), 6.40 (dd, 1H, J1′,2′ = 2.7 Hz, J1′,F-2′ = 22.3 Hz,H-1′), 6.24 (br.s, 2H, NH2), 5.34 (dd, 1H, J3′,2′ = 2.9 Hz, J3′,F-2′ = 17.9 Hz, H-3′), 5.17 (dd, 1H, J2′,F-2′ = 49.6 Hz, H-2′), 4.48 (dd, 1H, J4′,5′ = 5.5 Hz, J5′,5′′ = 12.0 Hz, H-5′), 4.40 (dd, 1H, J4′,5′′ = 3.7 Hz, H-5′′), 4.15-4.18 (m, 1H, H-4′). 13C NMR (CDCl3): δ 178.2 and 177.3 [(C=O, 2хСОС(СН3)3], 156.3, 154.5, 150.7, 118.0 (C-2, C-6, C-4, C-5), 140.4 (d, JC-8,F-2′ = 6.9 Hz, C-8), 92.9 (d, JC-2′,F-2′ = 194.4 Hz, C-2′), 83.4 (d, JC-1′,F-2′=16.9 Hz, C-1′), 81.26 (C-4′), 76.1 (d, JC-3′,F-2′ = 30.9 Hz, C-3′), 62.8 (C-5′), 39.0 and 38.9 [2xСОС(СН3)3], 27.27 and 27.1 [2xСОС(СН3)3]. 19F NMR (CDCl3): δ -197.56 (dt, F-2′). HRMS (EI): m/z calcd for C20H27ClFN5O5 (M+Na)+: 494.1582, found 494.1570.
Synthesis of 6-amino-2-chloro-9-(3¢,5¢-di-O-benzoyl-2¢-deoxy-2¢-fluoro-β-D-arabinofuranosyl)-9H-purine (20) and its α-anomer (21)
Method a1
To 2-chloroadenine (0.056 g, 0.32 mmol) in anhydrous 1,2-dimethoxyethane (12 ml) was added (0.038 g, 0.32 mmol) potassium tert-butoxide and the resulting mixture was stirred for 40 min under argon at room temperature and then anhydrous potassium bromide (0.058 g, 0.48 mmol) was added, and then suspension was stirred for 10 min and evaporated to dryness, coevaporated with anhydrous MeCN. Anhydrous MeCN (5 ml) was added to the residue and a suspension was stirred under argon at room temperature for 10 min, and then a solution of bromosugar 19 (0.116 g, 0.28 mmol) in MeCN (6.0 mL) was added dropwise at 00C to prepared potassium salt of the purine for 40 min. The reaction mixture was stirred under argon at room temperature for 18 h. Insoluble materials were removed by filtration and the solids were washed with CH2Cl2 (50 mL). The combined filtrate and washings were evaporated. The residue was chromatographed on silica gel, eluting with chloroform, CHCl3/EtOAc 7:1 and CHCl3/EtOAc/MeOH 7:1:0.1 to afford protected β-nucleoside 20 (0.063 g, 45%) as a foam and α-nucleoside 21 as a foam (0.022 g, 16%).
β-nucleoside 20. 1H NMR (CDCl3): δ 8.07-8.11 (2m, 10H, Bz), 8.05 (d, 1H, JH-8, F-2′ = 2.7 Hz, H-8), 7.44-7.67 (4m, 6H, Bz), 6.57 (dd, 1H, J1′,2′ = 2.2 Hz, J1′,F-2′ = 23 Hz,H-1′), 6.40 (br.s, 2H, NH2), 5.74 (dd, 1H, J3′,2′ = 2.3 Hz, J3′,F-2′ = 17.2 Hz, H-3′), 5.36 (dd, 1H, J2′,F-2′ = 49.8 Hz, H-2′), 4.81 (dd, 1H, H-5′), 4.78 (dd, 1H, H-5′′), 4.53-4.57 (m, 1H, H-4′). 13C NMR (CDCl3): δ 166.28 and 165.33 (C6H5CO), 156.39 (C-2), 154.44 (C-4), 150.75 (C-6), 140.41 (d, JC-8,F-2′ = 6.5 Hz, C-8), 134.3,133.5, 130.1, 129.9, 129.3, 129.4, 128.9, 128.7 (2Ph), 117.94 (C-5), 92.81 (d, JC-2′,F-2′ =191.48 Hz, C-2′), 83.62 (d, JC-1′,F-2′ = 17.0 Hz, C-1′), 81.32 (C-4′), 76.89 (d, JC-3′,F-2′ = 30.9 Hz, C-3′), 63.44 (C-5′). 19F NMR (CDCl3): δ -198.0 (dt, F-2′). HRMS (EI): m/z calcd for C24H19ClFN5O5 (M+Na)+: 534.0956, found 534.0951.
α-nucleoside 21. 1H NMR (CDCl3): δ 8.09-8.11 (m, 2H, Bz), 8.00 (s, 1H, H-8), 7.25-7.78 (m, 8H, Bz), 6.43 (d, 1H, J1′,2′ < 1.0 Hz, J1′,F-2′ = 14.4 Hz,H-1′), 6.25 (br.s, 2H, NH2), 6.16 (d, 1H, J2′,F-2′ = 49.0 Hz, H-2′), 5.79 (dm, 1H, J3′,F-2′ = 17.0 Hz, H-3′), 4.94-4.97 (m, 1H, H-4′), 4.70 (dd, 1H, H-5′), 4.68 (dd, 1H, H-5′′). 13C NMR (CDCl3): δ 166.27 and 165.15 (C6H5CO), 156.33 (C-2), 154.66 (C-4), 150.49 (C-6), 139.18 (s, C-8), 134.1, 133.4,129.9, 129.8, 129.3, 128.7, 128.6, 128.0 (2Ph), 119.1 (C-5), 96.6 (d, JC-2′,F-2′=188.5 Hz, C-2′), 89.49 (d, JC-1′,F-2′ = 36.0 Hz, C-1′), 81.3 (C-4′), 76.98 (d, JC-3′,F-2′ = 29.9 Hz C-3′), 63.48 (C-5′). 19F NMR (CDCl3): δ -188.0 (dt, F-2′). HRMS (ESI): m/z calcd for C24H19ClFN5O5 (M+Na)+: 534.0956, found 534.0949.
Method a2
To 2-chloroadenine (0.069 g, 0.39 mmol) in anhydrous 1,2-dimethoxyethane (16 ml) was added (0.045 g, 0.39 mmol) potassium tert-butoxide and the resulting mixture was stirred for 40 min under argon at room temperature and then anhydrous potassium bromide (0.075 g, 0.58 mmol) was added and then suspension was stirred for 10 min and evaporated to dryness. To a residue was added anhydrous MeCN (4.8 ml) and a suspension was stirred under argon at room temperature for 10 min, then anhydrous CH2Cl2 (2.4 ml) and a solution of bromide 19 (0.147 g, 0.33 mmol) in a mixture of MeCN/CH2Cl2 (8.4 mL, 2:1) was added dropwise at 00C to prepared potassium salt of the purine. The reaction mixture was stirred under argon at room temperature for 55 h. Insoluble materials were removed by filtration and the solids were washed with CH2Cl2 (35mL). The combined filtrate and washings were evaporated. The residue was chromatographed on silica gel, eluting with chloroform, CHCl3/EtOAc 7:1 and CHCl3/EtOAc/MeOH - 7:1:0.1 to afford protected β-nucleoside 20 (0.098 g, 55%) as a foam and α-nucleoside 21 (0.03 g, 17%) as a foam.
Synthesis of 6-amino-2-chloro-9-(2¢-deoxy-2¢-fluoro-β-D-arabinofuranosyl)-9H-purine (3)
Method a
A solution of nucleoside 20 (0.29 g, 0.57 mmol)in MeOH (14 mL) saturated at 0oC with ammonia was kept for 18 h at room temperature and then evaporated. To residue was added water (14 ml) and aqueous phase was extracted with EtOAc (3x14 ml), aqueous phase was evaporated and coevaporated with methanol. The residue was washed with ether (8 ml), crystallized from methanol to give nucleoside 3 (0.134 g, 78%). Mp. 230-2310С. 1H NMR (DMSO-d6): δ 8.22 (d, 1H, JH-8, F-2′ = 2.0 Hz, H-8), 7.84 (br.s, 2H, NH2), 6.28 (dd, 1H, J1′,2′ = 4.4 Hz, J1′,F-2′ = 14.1 Hz, H-1′), 5.92 (d, 1Н, J3′,3′-ОН = 5.2 Hz, 3′-OH), 5.23 (dt, 1H, J2′,F-2′ = 52.6 Hz, J2′,3′ = 4.2 Hz, H-2′), 5.04 (t, 1Н, J5′,5-ОН = 5.7 Hz, 5′-OH), 4.40 (dq, 1H, J3′,F-2′ = 18.8 Hz, H-3′), 3.8 (m, 1H, H-4′), 3.62 (m, 1H, J5′,4′ = 4.7 Hz, J5′,5′′ = 12.0 Hz, H-5′), 3.57 (dd, 1H, J5′′,4′ = 5.5 Hz, H-5′′). 13C NMR (DMSO-d6): δ 157.3, 153.8, 150.7, 140.52 (d, JC-8,F-2′ = 2.99 Hz, C-8), 117.9, 95.88 (d, JC-2′,F-2′ = 192.1 Hz, C-2′), 84.1 (d, JC-4′,F-2′ = 4.6 Hz, C-4′), 82.0 (d, JC-1′,F-2′ = 17.0 Hz, C-1′), 73.1 (d, JC-3′,F-2′ = 23.0 Hz, C-3′), 60.9 (C-5′). 19F NMR ((DMSO-d6): δ -198.38 (dt, F-2′). HRMS (ESI): m/z calcd for C10H11N5O3FCl(M+H)+: 304.0613, found 304.0597.
Method b
To solution of protected nucleoside 18 (0.046g, 0.097 mmol)in anhydrous methanol (0.7 mL) was added 0.35 ml 0.22 M sodium methoxide in methanol and prepared solution was stirring for 18 h at room temperature, then reaction mixture was neutralized with acetic acid and evaporated, coevaporated with a mixture of toluene:ethanol - 1:1 (20 ml). The residue was chromatographed on silica gel using for elution CHCl3,then CHCl3:MeOH-20:1 and 7:1 to afford nucleoside 3 as a white solid(0.022 g, 74%).
Synthesis of 1-methanesulfonyloxy-3-methylbutane (22) from 3-methyl-butanol
To solution 3-methyl-butanol (2.0 ml, 1.62g, 18.36 mmol)was added dropwise 18 ml 1M Li-tri-sec-borohydride in THF at 0 0C, the prepared solution was stirring for 30 min at room temperature, then 2.14 ml (27.54 mmol) mesyl chloride was added at 0 0C. The reaction mixture was stirred for 20 h at room temperature and poured out into water/ice (60 ml), water phase was extracted with chloroform (3 x100 ml). The organic extracts were washed with 1% aq. H2O2, water, dried over sodium sulfate, and evaporated. The residue was chromatographed on silica gel, using mixtures of 8:1, 5:1 and 3:1 hexane-EtOAc. 3-Methyl-butanol mesylate 22 was prepared as colorless syrup (2.1 g, 70%). 1H NMR (500 MHz, CDCl3) δ: 4.31 [t, 2Н, (CH3)2CHCH2CH2OMs], 3.05 (s, 3Н, -OSO2CH3), 1.78 - 1.87 [m, 1H, (CH3)2CHCH2CH2OMs], 1.68 (q, 2H, (CH3)2CHCH2CH2OMs], 0.99 (d, 6H, (CH3)2CHCH2CH2OMs]. 13C NMR (125 MHz, CDCl3) δ: 68.66 (-CH2OMs), 37.43 [(CH3)2CHCH2CH2-], 37.43 (OSO2CH3), 25.51 [(CH3)2CH-)], 22.27 [(CH3)2CH-].
Synthesis of 2-chloro-6-isopentylamino-9-(2¢-deoxy-2¢-fluoro-β-D-arabinofuranosyl)-9H-purine (23)
To solution of clofarabine 3 (0.132g, 0.435 mmol)in anhydrous DMSO (5.2 mL) was added potassium tert-butoxide (0.06 g, 0.52 mmol) and prepared solution was stirring for 15 min at room temperature, then mesyl derivative of 3-methyl-1-butanol (22) (0.27 ml, 0.231 g, 1.56 mmol) was added. The reaction mixture was stirred for 15 min at room temperature, and then 90 min at 89-90 0C, and, after cooling, a brown solution was diluted with water (3 ml) and aqueous phase was extracted with CHCl3 (3x40 ml). The combined organic extracts were evaporated, coevaporated with toluene (3x7ml). The residue was chromatographed on silica gel, eluting with CHCl3,then CHCl3:petroleum ether:MeOH - 30:6:1 and 6:4:1 to afford N6-isopentyl nucleoside 23 (0.05 g, 73%) as an amorphous powder taking into account of the recovery of the starting nucleoside 3 (0.08 g).Mp.184-186 0С.1H NMR (CD3OD): δ 8.24 (d, 1H, JH-8, F-2′ = 1.9 Hz, H-8), 6.41 (dd, 1H, J1′,2′ = 4.2 Hz, J1′,F-2′ = 15.4 Hz, H-1′), 5.16 (ddd, 1H, J2′,F-2′ = 52.2 Hz, J2′,3′ = 4.0 Hz, H-2′), 4.51 (ddd, 1H, J3′,4′ = 3.3Hz, J3′,F-2′ = 18.2 Hz, H-3′), 3.97 (m, 1H, H-4′), 3.85 (ddd, 1H, J5′,4′ = 3.8 Hz, J5′,F-2′ = 0.8 Hz, J5′,5′′ = 12.1 Hz, H-5′), 3.80 (dd, 1H, J5′′,4′ = 5.1 Hz, H-5′′), 3.58-3.62 [m, 2H, HNCH2CH2CH(CH3)2], 1.7-1.78 [m, 1H, CH2CH2CH(CH3)2], 1.57-1.61 [m, 2H, CH2CH2CH(CH3)2], 1.01 [br.s, 3H, CH(CH3)2], 1.34 [br.s, 3H, CH(CH3)2]. 13C NMR (CD3OD): δ 155.2, 154.5, 149.2, 117.5, (C-6, C-2, C-4, C-5), 139.78 (br.s, JC-8,F-2′ <2.0 Hz, C-8), 95.5 (d, JC-2′,F-2′ = 191.5 Hz, C-2′), 84.19 (d, JC-4′,F-2′ = 3.7 Hz, C-4′), 82.9 (d, JC-1′,F-2′ = 17.0 Hz, C-1′), 73.3 (d, JC-3′,F-2=24.3 Hz, C-3′), 60.7 (C-5′), 38.55 and 37.95 [CH2CH2CH(CH3)2], 25.5 [CH2CH2CH(CH3)2], 21.5 [CH2CH2CH(CH3)2]. 19F NMR(CD3OD): δ -199.5 (dt, F-2′). HRMS (ESI): m/z calcd for C15H21N5O3FCl (M+Na)+: 396.1215, found 396.1199.
Acknowledgments
This study was supported by grant from FOI «Chemical processes, reagents and technologies, bioregulators and bioorgchemistry», s/p «Chemical foundations of life activity processes» (Bioorgchemisrty 2.3.2.2) and partially with grant (Biologically active compounds 2.18).
References
- 1.Guinan M, Benckendorff C, Smith M, Miller G. (2020) . Recent Advances in the Chemical Synthesis and Evaluation of Anticancer Nucleoside Analogues,Molecules 25, 1-25.
- 2.Shelton J, Lu X, Hollenbaugh J, Cho J, Amblard F. (2016) . , Metabolism, Biochemical Actions, and Chemical Synthesis of Anticancer Nucleosides, Nucleotides, and Base Analogs, Chem. Rev 116, 14379-14455.
- 3.Parker W. (2009) Enzymology of Purine and Pyrimidine Antimetabolites Used in the Treatment of Cancer,Chem. , Rev 109, 2880-2893.
- 4.Robak T, Korycka A, Lech-Maranda E, Robak P. (2009) Current status of older and new purine nucleoside analogues in the treatment of lymphoproliferative diseases,Molecules. 14, 1183-1226.
- 5.Ewald B, Sampath D, Plunkett W. (2008) Nucleoside analogs: molecular mechanisms signing cell death,Oncogene. 27, 6522-6637.
- 6.Bonate P, Arhoud L, Cantrell W, Stephenson K, Secrist J et al. (2006) Discovery and development of clofarabine: a nucleoside analogue for treating cancer,Nature. Rev. Drug Discovery.5: 855-863.
- 7.Majda K, Lubecka K, Kaufman-Szymczyk A, Fabianowska-Majewska K. (2011) Clofarabine (2-Chloro-2´-Fluoro-2´-deoxyarabinofuranosyladenine) -. , Biochemical Aspects of Anticancer Activity,Acta Pol. Pharm. Drug. Res 68, 459-466.
- 8.Zhenchuk A, Lotfi K, Juliousson G, Albertioni F. (2009) Mechanism of anti-cancer action and pharmacology of clofarabine,Biochem. , Pharmacol 78, 1531-1359.
- 9.Daly M, Roth M, Bonnac L, Maldonado J, Xie J. (2016) Dual anti-HIV mechanism of clofarabine,Retrovirology. 13, 1-12.
- 10.Montgomery J, Shortnacy A, Clayton S, Riordan J, Secrist J. (1992) . Synthesis and Biological Activity of 2´-Fluoro-2-halo Derivatives of 9-β-D-Arabinofuranosyladenine,J. Med. Chem.35: 397-401.
- 11.Bauta W, Schulmeir B, Burke B, Puente J, Cantrell W. (2004) A New Process for Antineoplastic Agent Clofarabine,Org. Process Res. , Develop 8, 889-896.
- 12.Cen Y, Sauve A. (2010) Efficient Synthesis of clofarabine and gemcitabine from 2-deoxyribonolactone,Nucleosides, Nucleotides and Nucleic Acids. 29, 113-122.
- 13.Dostie S, Prevost M, Mochirian P, Tanveer K, Andrella N. (2016) Diastereoselective Synthesis of C2´-Fluorinated Nucleoside Analogues Using an Acyclic approach,J. , Org. Chem 81, 10769-10790.
- 14.Izawa K, Takamatsu S, Katayama S, Hirose N, Kozai S. (2003) An Industrial Process for Synthesizing Lodenosine (FddA),Nucleos. , Nucleot. Nucleic Acids 22, 507-517.
- 15.Fateev I, Antonov K, Konstantinova I, Muravyova T, Seela F. (2014) The chemoenzymatic synthesis of clofarabine and related 2-deoxyfluoroarabinofuranosyl nucleosides: the electronic and stereochemical factors determining substrate recognition byE.colinucleoside phosphorylases,Beilstein. , J. Org. Chem.10: 1657-1669.
- 16.Yamada K, Matsumoto N, Haykawa H. (2009) Stereoselective Synthesis of 2-Deoxy-2-Fluoroarabinofuranosyl-α-1-Phosphate and Its Application to the Synthesis of 2-Deoxy-2-Fuoroarabinofuranosyl Purine Nucleosides by a Chem-Enzymatic Method,Nucleosides, NucleotidesNucleic Acids. 28, 1117-1130.
- 17.Sivets G. (2016) Syntheses of 2-deoxy-2-fluoro-β-D-arabinofuranosyl purine nucleosides via selective glycosylation reactions of potassium salts of purine derivatives with the glycosyl bromide,Tetrahedron Lett. 57, 268-271.
- 18.Pankiewicz K, Krzeminski J, Watanabe K. (1992) Synthesis of 2´-β-Fluoro-and 3´-α-Fluoro-Substituted Guanine Nucleosides. Effects of Sugar Conformational Shifts on Nucleophilic Displacement of the 2´-Hydroxy and 3´-Hydroxy Group with DAST,J. Org. Chem 57, 7315-7321.
- 19.Pankiewicz K, Watanabe K. (1993) Synthesis of2´-β-fluoro-substituted nucleosides by a direct approach,J. , Fluor. Chem 64, 15-36.
- 20.Maruyama T, Takamatsu S, Kozai S, Satoh Y, Izawa K. (1999) Synthesis of 9-(2-Deoxy-2-fluoro-β-D-arabinofuranosyl)adenine Bearing a Selectively Removable Protecting Group,Chem. , Pharm. Bull 47, 966-970.
- 21.Sivets G, Kalinichenko E, Mikhailopulo I. (2006) Synthesis of C2´-β-Fluoro-substituted Adenine Nucleosides Via Pivaloyl Derivatives of Adenosine and 3'-Deoxyadenosine,Lett. , Org. Chem 3, 402-408.
- 22.Sivets G. (2018) Regio- and stereoselective syntheses of L-pentose derivatives from. , L-arabinose,Tetrahedron 74, 920-931.
- 23.Shao-Min W, Hong-Min L. (2008) Synthesis of Androrapholide Glucopyranoside and Selective Cleavage of O-Acetyl Groups in Sugar Moiety. , Chinese J. Chem 26, 343-347.