
Refractory Anaemia with Hyperoxalurea
Abstract
We report 2 cases of primary hyperoxalurea who presented with refractory anaemia, nephrolithiasis, renal failure warranting repeated renal transplantation in one of the cases. Renal biopsy of the patients revealed crystals of calcium oxalate in the tubules. The same crystals were also visualized in bone marrow biopsy which confirmed the diagnosis of systemic oxalosis. We conclude that Primary hyperoxalurea may rarely cause anemia secondary to calcium oxalate crystal deposition in the bone marrow.
Author Contributions
Academic Editor: Ying-Yong Zhao, Northwest University, Shaanxi 710069
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2015 Ayesha Ehsan, et al
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Introduction
Primary hyperoxalureas (PHs) are rare inborn errors of glyoxylate metabolism characterized by the overproduction of oxalate, predisposing to hyperoxalurea. Long-standing and unattended hyperoxaluria can progressively impair renal functions, and ultimately lead to renal failure.1, 2Once renal failure occurs, blood oxalate concentrations rise and precipitation occurs throughout the body. This stage is termed “oxalosis.” The common extra renal sites of oxalate deposition are the bones, bone marrow, blood vessels, central nervous system, peripheral nerves, retina, skin, and thyroid. 3 Renal Failure that ensues in due course of time in almost all the patients of oxalosis, is generally associated with anaemia. Deposition of oxalates in the bone marrow further aggravates anemia and other cytopenias may also be seen.4
Here we report the 2 cases of hyperoxalurea who presented with progressive anaemia not responding to therapy with iron, vitamin B12 or folic acid supplements. Their renal biopsies revealed characteristic oxalate crystals (whewellite crystals). Their bone marrow biopsy demonstrated systemic oxalosis due to underlying PH.
Case Report I
A 30 years old male, resident of Lahore was admitted for opinion regarding the necessity for regular hemodialysis after rejection of second renal transplant. Family history was suggestive of hereditary disease as two of his sisters had died of nephrocalcinosis leading to chronic renal failure at the age of 4 years. Consanguineous marriages in past many generations were also reported. Three cousins of the patient had recurrent stones with normal kidney functions. His past history revealed retention of urine at the age of 5 years due to stone in urethra. Patient presented again after 20 years in 2008 with retention of urine which was relieved by catheterization. Renal ultrasound and X ray KUB reported normal kidneys in both instances. Work up for stone formation was not undertaken at that time. The patient was put on hemodialysis and was planned for transplant which was undertaken in May 2009. After one month, follow up tests revealed Serum Creatinine to be 2.0 mg/dL. Renal Biopsy done in Sep 2009 revealed rejection of transplant. Hemodialysis was started and a second transplant was contemplated. Hemodialysis continued for 2 years till Dec 2011 when second transplant was carried out. A repeat renal biopsy was indicated for increasing serum creatinine. Again transplant rejection was evident on histology. Further investigations revealed Urinary oxalate excretion exceeding 40 mg/day (30 mg Oxalate/gram creatinine). His Blood investigations at the time bone marrow studies revealed bicytopenia with a Haemoglobin of 7.8g/dl, White blood cell count of 5.8 x 109/l; Platelet count of 120 x 109/l. Serum Creatinine was 3.6mg/dl. An attempt to correct the cytopenias by iron, vitamin B12 and folic acid supplements met with failure. No response to anemia could be elicited by adding erythropoietin to the treatment regimen. Renal and bone marrow biopsy were undertaken. Renal biopsy of the patient revealed crystals of calcium oxalate in the tubules (Figure 1). The same crystals were also visualized in bone marrow aspirate (Figure 2a and Figure 2b) and trephine biopsy (Figure 3).
Figure 1.Photomicrograph of renal tissue from case I with a preserved glomerulus and clusters of oxalate crystals in the tubules.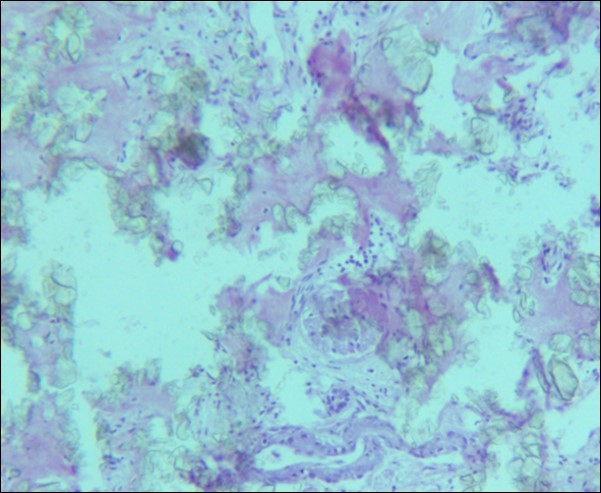
Figure 2a & b.Photomicrograph of bone marrow aspirate from case I revealing oxalate crystals interspersed in the bone marrow fragments and a foreign body giant cell.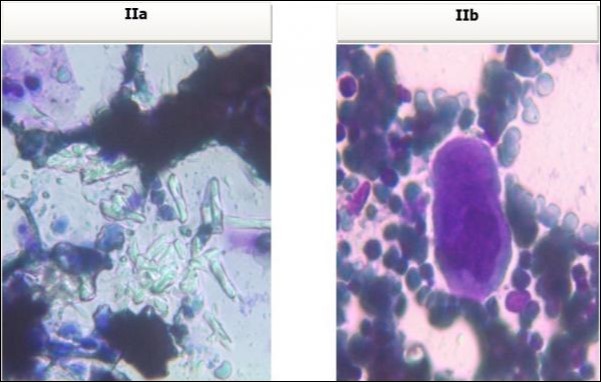
Figure 3.Photomicrograph of bone marrow core from case 1 showing radially arranged calcium oxalate crystals replacing haemopoietic tissue with invasion and destruction of bony trabeculae. Areas of necrosis and a cluster of hemosiderin laden macrophages is visible.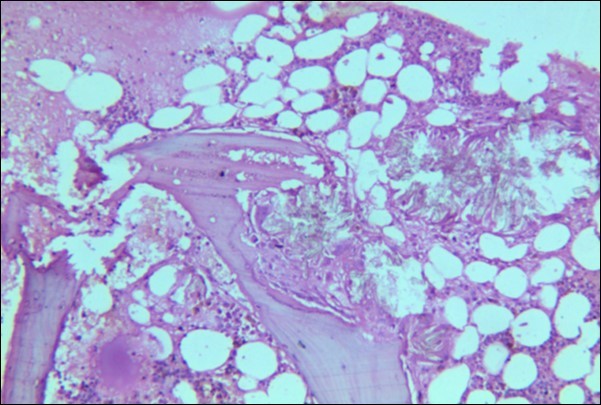
Case Report II
A 24 years old male, presented with the complaints of multiple recurrent renal stones bilaterally. He was on dialysis since last 6 months. His brother had died with chronic renal insufficiency secondary to nephrolithiasis. His routine investigations at the time bone marrow studies revealed anemia with a Haemoglobin of 6.6g/dl, White blood cell count of 5.3 x 109/l; Platelet count of 228 x 109/l. Serum Creatinine was very high 13.9mg/dl. X Ray Abdomen revealed multiple bilateral renal calculi and in addition gall bladder calculi. On ultrasound abdomen his right kidney was 10.5cm x 5.6 cm, had a cyst in the upper pole, multiple calculi were causing hydronephrosis. Left kidney was 8.5cm x 4.8 cm, with multiple calculi. Both kidneys showed increased echogenecity. Serum oxalate levels were 91.2 mg/dl (Normal Range 1-2.4 mg/dl). His Parathyroid MIBI scan was normal, but plasma Parathyroid hormone levels were 373 pg/ml (Normal Range 16 – 87pg/ml). Normochromic normocytic anaemia was present, with normal WBC and platelet count. The bone marrow aspirate was unsuccessful. The trephine biopsy revealed reduced cellularity, depressed erythropoeisis with partly replaced haemopoietic tissue with calcium oxalate crystals (Figure 4) which showed birefringence on polarized light (Figure 5).
Figure 4.Photomicrograph of bone marrow core from case2 showing Para trabecular arrangement of calcium oxalate crystals. Increased fat spaces and depressed erythropoiesis is evident.
Figure 5.Calcium oxalate crystals showing birefringence under polarized light.
Discussion
Systemic oxalosis can be a primary or a secondary disease. Primary hyperoxaluria Type 1 (PH 1) is due to the deficiency of glyoxylate aminotransferase and Type II due to that of glyoxylate reductase/D-glycerate dehydrogenase. PH 1 is an autosomal recessive disorder characterized by hyperoxaluria, calcium oxalate urinary lithiasis in childhood, nephrocalcinosis and renal failure which in turn leads to high blood oxalate levels and precipitation occurs throughout the body in the skin, blood vessels and joints. Secondary oxalosis occurs due to oxalate-rich diet, increased absorption or production of oxalate and reduced excretion as seen in renal failure. Variable degree of cytopenias or pancytopenia may accompany extensive oxalosis. Bone marrow aspirate may be unsuccessful due to extensive crystal deposition or accompanying fibrosis. Calcium oxalate crystals are well demonstrated on trephine biopsy with associated fibrosis and occasionally granulomas. They have a grey-yellow radial arrangement on hematoxylin and eosin staining and are birefringent under polarized light.4 Liver biopsy is the confirmatory test which demonstrate reduced glyoxylate aminotransferase activity (PH1). Molecular diagnosis, being non-invasive, is preferred, if available.5 An early diagnosis of oxalosis is of immense value, because at a stage when renal failure has not set in, proper management can arrest or at least delay the progress of disease. However, in patients who have already developed renal failure at the time of diagnosis, a combined liver and kidney transplantation offers the only salvation.6
Both the cases we report here showed prominent bone marrow infiltration by these crystals. Crystals were found imbedded in the bone marrow fragments. Occasional giant cell formation was also seen, however, granulomas were not visualised. The blood cytopenias/ anaemia were suggestive of the underlying pathology. Therefore we suggest that the diagnosis of primary hyperoxalurea be kept in mind when patients present with cytopenia and renal failure at an early age.
Conclusion:
Primary hyperoxalurea may be a rare cause of anemia secondary to calcium oxalate crystal deposition in the bone marrow.
References
- 1.Devonald M A, Karet F E. (2004) Renal Epithelial Traffic Jams and One-Way Streets. , J Am Soc Nephrol 15, 1370.
- 2.Kakkar N, Mittal D, Das S, John J M, Rajamanickam T. (2011) Bone marrow involvement in systemic oxalosis: A rare cause of leukoerythroblastic anemia. , Indian J Pathol Microbiol 54, 659-60.
- 3.Hassan K, Qaisrani J H, Qazi H, Naseem L, Zaheer H A et al. (2006) . Oxalosis in the Bone and Bone Marrow. International J Pathol; 4(2), 129-131.
- 4.Walter M J, Dang C V. (1998) Pancytopenia secondary to oxalosis in a 23-year-old woman. , Blood 91, 4394.
Cited by (5)
This article has been cited by 5 scholarly works according to:
Citing Articles:
Medicine theory and practice (2023) OpenAlex
Yuan-Yuan Chen, Dan-Qian Chen, Lin Chen, Jing-Ru Liu, N. Vaziri et al. - Journal of Translational Medicine (2019) Semantic Scholar
Journal of Translational Medicine (2019) OpenAlex
Journal of Translational Medicine (2019) Crossref
Shi-Xing Ma, You-Quan Shang, Huan-Qiao Zhang, Wei Su - Journal Of Nephrology Advances (2018) Semantic Scholar
Journal Of Nephrology Advances (2018) OpenAlex
Journal of Nephrology Advances (2018) Crossref