
Abstract
N-acetylaspartylglutamate (NAAG) is the highest concentration dipeptide present in brain. It is found primarily in neurons but its function is unclear. NAAG is synthesized by neurons from N-acetylaspartate and glutamate (Glu), maintained at mM concentrations and is released non-synaptically to extracellular fluid (ECF). NAAG is a non-excitatory form of Glu, and is targeted to the metabotropic group II Glu receptor 3 (mGluR3) on the surface of astrocytes. After docking with the receptor, Glu is released by the action of NAAG peptidase. Previously, it was shown for the first time that an NAAG-peptidase inhibitor reduced global cerebral blood flow (CBF) in mouse brain but did not affect their physical performance. Recently, it has been demonstrated that there are two separate systems involved in neurovascular coupling by astrocytes, one is a rapid focal phasic response providing energy for stimulation-induced neuronal activity, and the other a slower global tonic response providing energy for ongoing metabolic activities. Many neurovascular coupling mechanisms are known that regulate phasic changes in CBF, but how the brain accomplishes tonic control is unknown. In this paper we bring together two separate lines of inquiry, the decades’ long search for the function of NAAG, and the more recent search for the mechanism of tonic neurovascular control. Herein, we present evidence that NAAG is the neurovascular coupling agent that regulates tonic changes in CBF via the astrocyte mGluR3-NAAG peptidase connection.
Author Contributions
Academic Editor: Ramesh C Gupta, Visiting Professor, National Institute of complementary Medicine (NICM) University of Western Sydney Australia
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2016 Morris H Baslow, et al
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Introduction
The Question of the Function of NAAG
The presence of high levels of N-acetylaspartate (NAA) and N-acetylaspartylglutamate (NAAG) in the mammalian brain, and of most of the enzymes that synthesize and hydrolyze them were discovered more than six decades ago. At that time it was also noted that these substances and their anabolic and catabolic enzymes were highly compartmentalized with most NAA and NAAG and their anabolic enzymes present in neurons, and their catabolic enzymes present in macroglia. These early findings have been reviewed 1, 2. Subsequently, in a review of the properties of metabotropic glutamate receptors (mGluR’s) found in brain, it was reported that of the eight members of the three groups of mGluR’s, that NAAG was a selective agonist for the group II mGluR3 receptor 3. At the same time, two papers were published, one that demonstrated for the first time that the enzyme that hydrolyzed NAA was only expressed in oligodendrocytes 4, and the other that showed that the enzyme that hydrolyzed NAAG was expressed “exclusively in astrocytic glial cells” 5. The “nagging” question of the function of NAAG was considered in 1997 6 and revisited in 2015 7.
The hypothesis that NAAG was intimately involved in neuron-astrocyte communication was first suggested in 1999 5 and it was speculated that NAAG, via the action of its substrate-specific astrocytic peptidase, “may be an important mediary of neuronal-glial communication”. The expanded NAAG functional hypothesis was a logical step in the evolution of the concept in that it considered the entire NAA-NAAG metabolic sequence as a unique linked tri-cellular system and stated that “NAAG in the CNS may have a …primary role” in “neuronal-glial cell-specific signaling and communication” 8. In 2005 it was observed for the first time that by inhibiting NAAG peptidase in vivo, the astrocytic enzyme that hydrolyzes NAAG, that there was a prolonged global reduction in the proton magnetic resonance blood oxygen level-dependent (BOLD) signal indicating a reduction in global cerebral blood flow (CBF), but with little or no effect on physical activity 9. In 2006 the hypothesis was expanded to suggest that NAAG functioned “to control focal or regional hyperemia” by stimulating astrocytes to synthesize and release second messengers to the vascular system via cyclooxygenase-1 (COX-1) synthesized prostaglandins 10.
A number of recent studies support aspects of the original hypothesis. In 2013, it was demonstrated that the mGluR3 receptor was the predominant mGluR receptor present in mature murine and human astrocytes and that other mGluRs were very low or absent 11. Thus, not only is NAAG uniquely targeted to the mGluR3 receptor, but it may be the only mGluR receptor on the surface of mature astrocytes, the cells that are integral to regulating near-field CBF 12. Additional support for the hypothesis was developed in 2015, when the presence of a neuronal efflux transporter that transports both NAA and NAAG into ECF by a non-synaptic mechanism was reported 13, thus providing a possible mechanism for their continuous release to oligodendrocytes and astrocytes respectively.
The Bimodal Nature of Neurovascular Coupling
The brain exhibits a remarkable feature in that there is a high degree of functional specialization in a relatively small organ. As a result there are many different small neuronal “neighborhoods”, independent of neuron type or connectivity, that require different temporal allocations of CBF in order to supply sufficient quantities of glucose (Glc), O2 and nutrients, and also to serve as a sink for waste products CO2, H2O and generated heat. This is accomplished by an intricate system of chemical feedback signals between neurons, astrocytes and the vascular system. This cellular association has been termed the “neurovascular unit” and the crosstalk between the cells called neurovascular coupling (NVC) 14, 15. In this process, glutamate (Glu) plays an important role in activating astrocytes to initiate signaling to the vascular system 12. In 2015, it was reported that there were two different types of NVC, one rapid and phasic in response to abrupt changes in neuron synaptic activity, inducing astrocyte Ca2+ oscillations and eliciting immediate vascular responses. The other, slow and tonic and independent of regional changes in neuronal synaptic activity, using resting intracellular Ca2+ and continuous release of COX-1 generated second prostaglandin messengers 16. Many neurovascular coupling mechanisms are known that can regulate phasic changes in CBF 14, 15, but how the brain accomplished tonic control of CBF was reported to be unknown. In this analytical review, we bring together evidence of a signaling mechanism that matches the criteria for tonic regulation CBF and suggests that the function of neuronal release of NAAG in brain is to regulate tonic control of CBF.
Discussion
The Tri-Cellular Metabolism of NAA and NAAG Metabolism
NAA and NAAG are among the highest concentration amino acids and dipeptides present in brain, and are almost exclusively found in neurons. Within neurons, the ratio of NAAG to NAA is lowest in gray matter (GM) and highest in white matter (WM) 1. NAA is synthesized from L-aspartate (Asp) and acetyl Co-enzyme A (AcCoA) by NAA synthase 17 with Glc the source of acetate (Ac) in AcCoA. NAA is the only known precursor of NAAG, a non-excitatory form of Glu synthesized from NAA and Glu in neurons by NAAG synthase 18. Importantly, because of their genesis, the rates of synthesis of both NAA and NAAG always reflect the rate of neuronal Glc oxidation, with about 1 molecule of NAAG synthesized for every 320 molecules of Glc oxidized 19. This is shown in equation 1.
Eq 1. (Neurons, NAA and NAAG synthases)
11520 ADP + 11520 P + 320 Glc + 1920 O2 + 11 Asp + 1Glu 1920 CO2 + 1920 H2O+11520 ATP + 10 NAA + 1 NAAG
Most neurons in brain synthesize NAA and NAAG and store large quantities of both substances. However, neurons cannot catabolize either of these substances. For their catabolism, they are exported to extracellular fluid (ECF) 20. NAA is targeted to oligodendrocytes where it is hydrolyzed by aspartoacylase (ASPA) liberating Ac and Asp 21, (Eq 2), and NAAG is targeted to the mGluR3 receptor on the astrocyte surface where the Glu is cleaved by NAAG peptidase 22, (Eq 3.).
Eq 2. (Oligodendrocytes, ASPA)
NAA Ac + Asp
Eq 3. (Astrocytes, NAAG peptidase)
NAAG NAA + Glu
NAA is also a byproduct of astrocyte NAAG hydrolysis but astrocytes cannot further metabolize it. For its catabolism it must be liberated to ECF and hydrolyzed by oligodendrocyte ASPA. The unique tri-cellular metabolism of NAA and NAAG with two synthetic and two hydrolytic enzymes distributed between three cell types, and the NAAG-mGluR3-NAAG peptidase Glu release mechanism on the astrocyte surface has been called the "operating system" of the brain. This is because failure of several parts of the system in humans has been observed to lead to abnormal brain function 23. The group II mGluR3 is also unique among mGluR’s in that there is an astrocyte-targeted neuron-dedicated neurotransmitter (NAAG) and an associated specific enzyme (NAAG peptidase) for its hydrolysis 24. The mGluR3 is a G-protein Gi/Go bound receptor negatively coupled to adenylate cyclase that does not trigger Ca2+ increases in astrocytes, thus excluding its
involvement in rapid synaptic events 11. A cartoon showing this association is presented in Figure 1.
Figure 1.Cartoon of proposed channel-like complex showing the association of NAAG, the mGluR3 receptor and NAAG peptidase on the astrocyte surface. After docking, Glu is released to the cytosol by NAAG peptidase, activating COX-1 and secondary release of prostaglandins to the vascular system inducing a hyperemic response and increasing the supply of oxygen and glucose. Extrinsic agonists, antagonists and NAAG peptidase inhibitors that mimic NAAG structure interfere with access of natural NAAG to the receptor-enzyme complex, disrupting this dynamic neurovascular energy supply mechanism (24).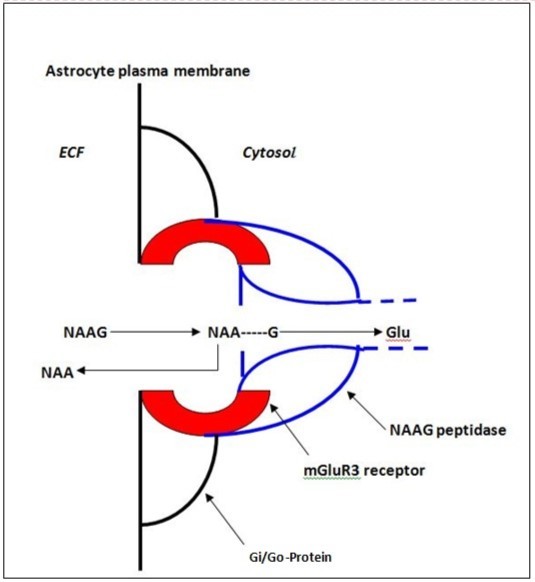
Mechanism of export of NAA and NAAG to ECF is non-synaptic and may also be associated with membrane depolarization
Both NAA and NAAG are released to ECF upon neuron depolarization. This has been observed for NAA in rat microdialysis studies where neurons were depolarized by K+25, 26 and for NAA and NAAG in a rat brain slice superfusion study where depolarization was by electrical stimulation at 15 Hz, 20mA for 3 min and NAA and NAAG were observed to be released at the same relative % rate 27. The release of NAA and NAAG is likely primarily non-synaptic since neurons do not express ASPA or NAAG peptidase that would be required for their synaptic catabolism and recycling. In many cartoon figures published over past decades that show the proposed synaptic release of NAAG, the NAAG is always depicted as leaving the synapse to be hydrolyzed by astrocyte NAAG peptidase. The neuron membrane ATP-binding cassette subfamily C, member 5 (ABCC5) has recently been reported to be an ATP-dependent non-synaptic efflux transporter for both NAA and NAAG 13 and is presently the only specific neuron NAA and NAAG exporting mechanism that has been identified. In mice lacking the gene for ABCC5, NAAG accumulated and was found to be 2.4 times higher in brain. If retained within neurons which represent about 50% of brain volume, this value would be doubled. While NAA and NAAG are not required for neuron plasma membrane depolarization or synaptic signaling as evidenced by gene knockout (KO) studies 7, neuron plasma membrane depolarization with or without associated synaptic signaling can trigger NAA and NAAG release.
NAAG catabolism can influence focal blood flow by releasing Glu, inducing vascular arteriole relaxation and corresponding lumen expansion
Neurons release their NAAG to ECF at a rate of about 6%/h and it is targeted to the astrocyte mGluR3 receptor-NAAG peptidase complex where the Glu moiety is cleaved. Glu can initiate astrocyte release of second messengers to vascular endothelial cells of capillaries and arterioles affecting CBF 28, and in a cortical slice experiment in-vitro, it was found that activation of isolated astrocyte-arteriole preparations resulted in a slow increase in the cross-sectional volume of arterioles 12. Relaxation of arterioles produces a transient increase in focal blood flow and as a result an increase in the ratio of oxygenated to deoxygenated blood, a change that can be measured using magnetic resonance imaging (MRI), as a positive BOLD difference. MRI measures a water signal and paramagnetic deoxyhemoglobin reduces this signal. Thus, as deoxyhemoglobin content is reduced by increased CBF, the BOLD signal rises. Stimulus driven or resting state temporal changes in BOLD in brain areas of interest can be identified using “functional” or time-based MRI (fMRI). The special case of NAAG, an inert carrier of Glu, and a dedicated physiological agonist of the mGluR3 receptor, argued for an important role for the complex NAAG-mGluR3-NAAG peptidase system in neuron-astrocyte communication and CBF regulation. Since mGluR3 may be the only mGluR receptor expressed in mature mouse and human astrocytes 11, this further supported the notion of its special role in astrocyte stimulation and neurovascular coupling.
Identification of Separate Mechanisms for Long-Term Tonic and Short-Term Phasic Increases in Focal Blood Flow
Neurons have two distinct requirements for energy. One for long-term energy supplies to meet the needs of ongoing housekeeping tasks such as maintenance of osmotic pressure, active transport of amino acids and other metabolites against gradients, synthesis of thousands of metabolic intermediates, and for building proteins and cellular structures. The other is to carry out rapid information processing via synaptic frequency-encoded signaling. These requirements are separate and can be observed by blocking specific astrocyte-vascular system signaling pathways to decrease blood flow and then superimposing stimulation-induced neuron firing to rapidly increase the need for immediate supplies of Glc and oxygen 16. These two modes of neurovascular coupling for energy supply have been termed tonic and phasic respectively 16 and they can be observed using changes in BOLD for both small “resting state” tonic fluctuations, as well as for large phasic stimulation-induced rapid changes in focal blood flow.
Evidence that NAAG, A Dedicated Agonist at the mGluR3 Astrocyte Receptor Functions as the Tonic Neurovascular Coupling Agent
Although discovered decades ago, the function of neuron synthesized NAAG remained a mystery. In an anesthetized mouse study, it was observed that a specific inhibitor of the mGluR3-associated NAAG peptidase, 2-(phosphonomethyl) pentanedioic acid (2-PMPA), resulted in a prolonged global reduction in BOLD of about 3% while vital signs were maintained, but had no effect on physical activity of awake mice in rotarod testing over 24 h 9. This was the first evidence that NAAG was a global neurotransmitter involved in long-term tonic regulation of blood flow, but not in short-term stimulation-induced changes to meet rapid phasic requirements for increased energy. COX-1 is involved in the production of prostaglandins which, when released by astrocytes induce a hyperemic response 29. In this study using a COX-1 inhibitor blocking second prostaglandin messengers to the vascular system from astrocytes, vibrissal stimulation in mice was still able to induce a phasic response. As a check, homozygous COX-1-null mice in this same study which did not synthesize prostanoids were observed to still retain this phasic response to stimulation. These authors concluded that phasic responses were clearly different from tonic responses and that the prostanoid products of COX-1 were critical in maintaining resting cerebrovascular tone. The studies also demonstrated that the tonic regulation of blood flow based on the regular release of prostaglandins is decoupled from phasic responses. Evidence that neuronal and/or astrocyte generated nitric oxide (NO) is an important mediary of phasic coupling was demonstrated in mice where it was shown that the phasic CBF response in the somatosensory cortex to vibrissal stimulation was decreased by half in the presence Nw-nitro-L-arginine methyl ester, an inhibitor of NO synthase 30. Since the synthesis of NAAG is directly connected to the rate of neuron Glc oxidation (Eq. 1), and it is released to the mGluR3-NAAG peptidase complex on the astrocyte surface for its catabolism resulting in liberation of Glu that can activate astrocytes to induce COX-1 activity and the export of prostaglandins, we propose that NAAG is responsible for long-term tonic neurovascular coupling.
Summary of the Evidence that NAAG is the Tonic Regulator of global CBF
While additional functions are possible, the evidence that NAAG is the neurovascular coupling agent that regulates tonic changes in CBF via the astrocyte mGluR3-NAAG peptidase connection is as follows. NAAG is synthesized by neurons as a function of the rate of Glc oxidation and is released to ECF continuously and targeted to the mGluR3 receptor on the astrocyte surface. After docking with the mGluR3 receptor, the action of mGluR3-coupled NAAG peptidase cleaves the Glu activating the astrocyte to communicate with the vascular system via release of prostaglandins, the outcome of which is to influence arteriole dilation and associated CBF. Inhibiting the action of NAAG peptidase rapidly and dramatically increases NAAG levels in ECF 31 and also reduces global CBF 9. The evidence in support of the role of NAAG as a tonic regulator of CBF, and the relationship between NAAG release and astrocytes shown in Table 1.
Table 1. Summary of evidence that NAAG is a neurotransmitter and regulator of tonic CBF| Neurons are the primary source of NAAG in the brain; NAAG can only be synthesized from NAA | ||||||||
| NAAG is a non-excitatory form of Glu | ||||||||
| NAAG is released to ECF continuously, and also upon neuron depolarization | ||||||||
| NAAG is released to ECF extra-synaptically | ||||||||
| NAAG is specifically targeted to the mGluR3 receptor on the astrocyte surface | ||||||||
| The only mGluR receptor expressed in mature astrocytes is mGluR3 | ||||||||
| The only enzyme that can hydrolyze NAAG, NAAG peptidase, is associated with the Gi/Go-protein - | ||||||||
| -coupled astrocytic mGluR3 receptor | ||||||||
| Inhibiting astrocyte NAAG peptidase results in the rapid buildup of NAAG in ECF and a global - | ||||||||
| -decrease in CBF | ||||||||
| mGluR3 activation stimulates astrocytes but does not trigger rapid Ca2+ increases- | ||||||||
| -The products of NAAG hydrolysis are Glu and NAA | ||||||||
| Glu activates astrocytes to liberate gliotransmitter prostaglandins via COX-1 and communicate with | ||||||||
| -the vascular system | ||||||||
| The continuous release of NAAG to ECF by neurons is associated with resting state or tonic | ||||||||
| -neurovascular coupling, but stimulation-induced release of NAAG may also participate in | ||||||||
| -prolongation of phasic neurovascular coupling | ||||||||
| In lower forms, the absence of NAA and NAAG, NAAG peptidase or mGluR3 is not lethal. Neurons are - | ||||||||
| -myelinated and can signal, and the animals appear to be little affected. However, in a single human | ||||||||
| -case where NAA and NAAG are absent there are profound developmental defects. | ||||||||
The interaction of NAAG with astrocytes and its control of tonic blood flow is graphically illustrated in Figure 2. In this cartoon we see that tonic control of focal CBF using NAAG is non-synaptic and is accomplished on an individual neuron basis. Thus, tonic control of CBF is a function of the collective needs of neighborhoods of individual neurons, each of which exports NAAG molecules at a rate that is a tied to its individual rate of Glc oxidation.
Figure 2.Cartoon showing a single neuron and the non-synaptic release of NAAG to ECF as a mediator of astrocyte-induced slow tonic changes in focal blood flow using resting intracellular Ca2+ and release of COX-1 synthesized prostaglandin second messengers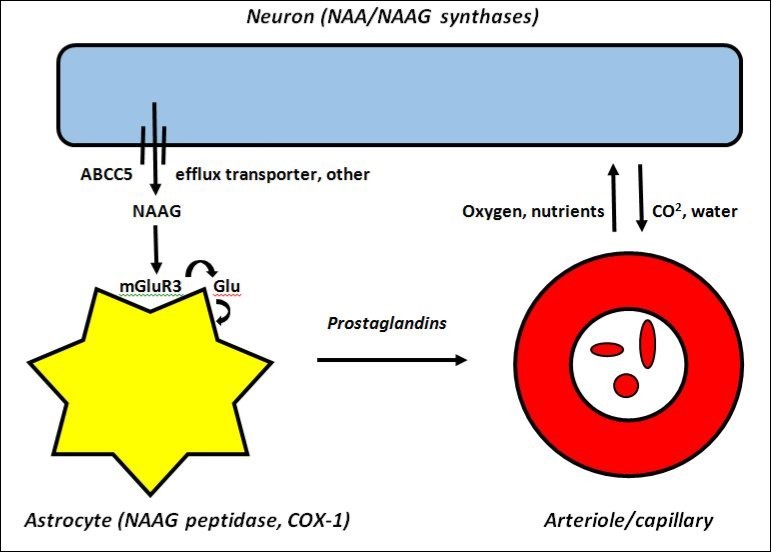
The Relationship Between Tonic and of CBF. Phasic Mechanisms for Regulation
We propose that the difference between tonic and phasic CBF interactions is that tonic interactions involve populations of single neurons continuously liberating a form of non-excitatory Glu (NAAG) to astrocytes, whereas phasic responses are generated by synaptic events between many neurons resulting in synaptic-released clouds of K+ and excitatory Glu as well as other substances leaked to ECF. The slow tonic system uses astrocyte COX-1 to generate a continuous release of prostaglandins to influence CBF whereas the rapid phasic system is more complex, having various temporal components as well as many different mechanisms that can be used to influence changes in CBF 32. Early phasic triggers may be due to the effect of synaptic neuron-released K+ or free Glu on astrocytes or other neurons resulting in synthesis and liberation of NO, or by direct neuronal or astrocytic signaling to arteriole smooth muscles. Later responses may include tonic NAAG-mGluR3 interactions producing Glu and then secondary astrocytic messengers to the vascular system released by the Glu-activated astrocytes. In Table 2 we summarize and compare the proposed mechanisms regulating tonic and phasic control of CBF.
Table 2. Genesis of neurovascular tonic and phasic coupling| Component | Tonic coupling | Phasic coupling |
|---|---|---|
| Trigger | rate of Glc oxidation | rate of synaptic firing |
| Timeframe | minutes | seconds |
| Number of neurons | each as individual | 2 or more, synaptically joined |
| Neurotransmitter | NAAG (bound Glu) | K +, Glu (free), other |
| Transmitter source | ABCC5 efflux transporter, other | synaptic cleft release |
| Astrocyte receptors | mGluR3 | K+, Glu, other |
| Astrocyte enzyme | NAAG peptidase | - |
| Astrocyte product | Glu | - |
| Astrocyte Ca2+ | resting Ca2+ | Ca2+ oscillations |
| Glio-message activator | COX-1 | NO synthase, other |
| Vascular messengers | prostaglandins | NO, other |
| Vascular target | capillaries, arterioles | capillaries, arterioles |
| CBF | slow changes in CBF (min) | rapid changes in CBF (s) |
| Glc | steady-state maintained | rapid depletion, rapid resupply |
| Oxygen | steady-state maintained | rapid depletion, rapid resupply |
| ATP energy stores | steady-state maintained | rapid depletion, rapid re-synthesis |
| Response measure | resting BOLD fluctuations | rapid large focal BOLD changes |
| Response inhibitors | COX-1 inhibitors | Synaptic firing inhibitors |
| NAAG peptidase inhibitors | NO synthase inhibitors |
In Figure 3 the proposed interaction of the NAAG system with additional elements that control phasic changes in blood flow is presented where it is shown that multiple neurons are involved and that synaptic firing is a key component.
Figure 3.Cartoon showing both (A) non-synaptic release of NAAG as a mediator of astrocyte-induced slow tonic changes in blood flow using resting intracellular Ca2+ and (B) synaptic release of K+ and free Glu as mediators of astrocyte-induced rapid phasic changes in blood flow using intracellular Ca2+ oscillations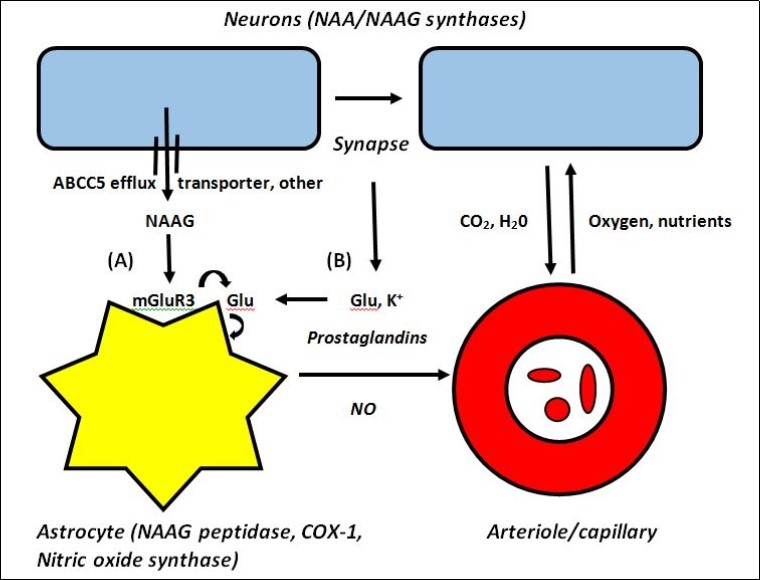
Conclusions
The function of NAAG is to regulate slow tonic changes in CBF needed for maintenance of neuronal integrity
After decades of searching for an answer to the question of the function of NAAG, recent findings related to the dual nature of brain neurovascular coupling have provided for a reasonable hypothesis; that NAAG is the tonic global regulator of CBF to meet ongoing focal requirements of individual neurons for energy to maintain long-term neuronal integrity. It is dependent on safe delivery and release of excitatory Glu to astrocytes in the form of NAAG, and on the subsequent stimulation of COX-1 activity to produce prostaglandins for continuous release to capillaries and arterioles to regulate their degree of relaxation and thus change focal CBF. This is a relatively simple system that is metabolically separate from phasic responses needed for the large influx of energy required by neurons to support rapid changes in the rate of neuronal firing. Neuronal NAAG, its dedicated astrocyte receptor mGluR3, and its dedicated astrocyte catabolic enzyme NAAG peptidase, are rarely mentioned or considered in the numerous reviews of neurovascular coupling mechanisms 32, 33. In one case where the role of mGluR3 was considered, it was concluded that “it is unlikely that mGluR3 directly mediates NVC” 14. In this paper, we show that contrary to such a conclusion, the NAAG system is a major component of NVC at all times and is involved in regulating the constant needs of neurons for energy to maintain cellular integrity even in the absence of synaptic firing and evoked astrocytic Ca2+ signals. Because the tonic system is independent of synaptic firing 16, it follows that it may be the most important regulator of CBF in GM during sleep when brain activity is reduced 33, as well as the primary regulator of CBF in WM, via release to astrocytes at nodes of Ranvier, where there are few synapses and NAAG is found in highest concentration 20.
Failure of the NAAG System may be Involved in Various Brain Neuropathies
We present evidence that NAAG functions as the global regulator of tonic regional blood flow to meet the ongoing housekeeping energy needs of neurons, estimated to be about 50% of their total energy requirements 16. While NAAG release and its tonic function is independent of synaptic events, its generation is Glc oxidation-dependent and therefore cannot be separated from, and must also contribute to changes in blood flow during intense short-term energy requiring synaptic phasic events. The complex nature of its putative role in regulation of blood flow to neurons, with the impact of the complete absence of NAAG in lower forms seemingly subtle and apparently only affecting certain behaviors 34, is of interest. However, its absence in a singular human case is associated with profound deleterious changes, suggesting that changes in NAAG metabolism may play an important role in the generation of some human neurological and psychopathological conditions. Although tonic and phasic elements of CBF cannot be completely separated, based on the evidence presented in this review we believe that the NAAG system is the primary regulator of resting state CBF, and a major contributor to resting state BOLD measurements. These measurements are characterized by modest focal fluctuations in BOLD and are associated with relatively low levels of synaptic activity. One possible outcome of the failure of this tonic system could be a chronic mismatch between energy needs and the availability of oxygen and Glc, thus affecting the rate of ATP regeneration and steady-state levels of ATP, and therefore the ability of neurons to rapidly repolarize and be able to transmit a full range of meaningful frequency-encoded messages 35. Chronic hypoxia has been linked to episodes of hallucinations and deficits in language production, cognition and memory. Currently, use of drugs that affect the tri-cellular metabolism of NAAG have been tested for their effects in animal models of disease, and as a result several have been advocated for possible treatment of human brain diseases and conditions including hypoxic and traumatic brain injury, stroke, diabetic neuropathy, amyotrophic lateral sclerosis, multiple sclerosis, Alzheimer’s disease and schizophrenia 36, 37, 38. Given a rationale for how NAAG may impact neuron physiology and their ability to communicate, a more focused approach to therapeutic interventions may now be possible.
Abbreviations Used:
Ac, acetate; AcCoA, acetyl Co-enzyme A; Asp, aspartate; ASPA, aspartoacylase; BOLD, blood oxygenation-level dependent; CBF, cerebral blood flow; COX-1, cyclooxygenase-1; ECF, extracellular fluid; fMRI, functional magnetic resonance imaging; Glc, glucose; Glu, glutamate; mGluR3, GM, gray matter; metabotropic Glu receptor 3; KO, knockout; MRI, magnetic resonance imaging; NAA, N-acetylaspartate; NAAG, N-acetylaspartylglutamate; NVC, neurovascular coupling; 2-PMPA, 2-(phosphonomethyl) pentanedioic acid; WM, white matter
References
- 1.D L Birken, W H Oldendorf. (1989) N-acetylaspartic acid: a literature review of a compound prominent in 1H-NMR spectroscopic studies of brain. , Neurosci. Biobehav. Rev 13, 23-31.
- 2.M H Baslow. (1997) A review of phylogenetic and metabolic relationships between the acylamino acids, N-acetyl-L-aspartic acid and N-acetyl-L-histidine in the vertebrate nervous system. , J. Neurochem 68(4), 1335-1344.
- 3.Schoepp D D, Jane D E, J A Monn. (1999) Pharmacological agents acting at subtypes of metabotropic glutamate receptors. , Neuropharmacol 38, 1431-1476.
- 4.M H Baslow, Suckow R, Sapirstein V, B L Hungund. (1999) Expression of aspartoacylase activity in cultured rat macoglial cells is limited to oligodendrocytes. , J. Mol. Neurosci 13, 47-53.
- 5.U V Berger, Luthi-Carter R, L A Passani, Elkabes S. (1999) Glutamate carboxypeptidase II is expressed by astrocytes in the adult rat nervous system. , J. Comp. Neurol 415, 52-64.
- 6.J T Coyle. (1997) The nagging question of the function of N-acetylaspartylglutamate. , Neurobiol. of Dis 4, 231-238.
- 7.M H Baslow. (2015) An answer to “The nagging question of the function of N-acetylaspartylglutamate “(NAAG). , Neurosci. Comm 2, 844-10.
- 8.M H Baslow. (2000) Functions of N-acetyl-L-aspartate and N-acetyl-L-aspartylglutamate in the vertebrate brain: Role in glial cell-specific signaling. , J. Neurochem 75, 453-459.
- 9.M H Baslow, Nowak K, B L Hungund, D N Guilfoyle.VV Dyakin.(2005) 2-PMPA, a NAAG peptidase inhibitor, attenuates the BOLD signal in brain of anesthetized mice: Evidence of a link between NAAG release and hyperemia. , J. Mol. Neurosci 26(3), 1-16.
- 10.M H Baslow, D N Guilfoyle. (2006) Functions of N-acetylaspartate and N-acetylaspartylglutamate in brain: Evidence of a role in maintenance of higher brain integrative activities of information processing and cognition. , Adv. Exptl. Med. and Biol 576, 95-112.
- 11.Sun W, McConnell E, Pare J-F, Xu Q. (2013) Glutamate-dependent neurological calcium signaling differs between young and adult brain. , Science 339, 197-200.
- 12.Zonta M, M C Angulo, Gobbo S, Rosengarten B, K A Hossmann et al. (2003) Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. , Nature Neurosci 6, 43-50.
- 13.R S Jansen, Mahakena S, M de Hass, Borst P, Wetering K van de. (2015) ATP-binding cassette subfamily C member 5 (ABCC5) functions as an efflux transporter of glutamate conjugates and analogs. , J. Biol. Chem 290(51), 30429-30440.
- 14.K M Dunn, M T Nelson. (2014) Neurovascular signaling in the brain and the pathological consequences of hypertension. , Am. J. Physiol. Heart Circ. Physiol. 306: H 1 - H 14.
- 15.Dormanns K, Disseldorp E M J van, R G Brown, David T. (2015) Neurovascular coupling and the influence of luminal agonists via the endothelium. , J. Theoret. Biol 364, 49-70.
- 16.D G Rosenegger, Tran C H T, I Wamsteeker Cusulin J, Gordon G R. (2015) Tonic local brain blood flow control by astrocytes independent of phasic neurovascular coupling. , J. Neurosci 35, 1346-13474.
- 17.Wiame E, Tyteca D, Pierrot N, Collard F, Amyere M et al. (2010) Molecular identification of aspartate N-acetyltransferase and its mutation in hypoacetylaspartia. , Biochem J 425, 127-136.
- 18.Becker I, Lodder J, Gieselmann V, Eckhardt M. (2010) Molecular characterization of N-acetylaspartylglutamate synthetase. , J. Biol. Chem 285, 29516-29164.
- 19.M H Baslow, D N Guilfoyle. (2007) Using proton magnetic resonance imaging and spectroscopy to understand brain "activation". , Brain and Language 102, 153-164.
- 20.M H Baslow, D N Guilfoyle. (2009) Are astrocytes the missing link between lack of brain aspartoacylase activity and the spongiform leukodystrophy in Canavan disease?. , Neurochemical Research 34(9), 1523-1534.
- 21.Bitto E, C A Bingman, G E Wesenberg, J G McCoy, G N Philllips. (2007) Structure of aspartoacylase, the brain enzyme impaired in Canavan disease. , PNAS 104, 456-461.
- 22.Sacha P, Zamecnik J, Barinka C. (2007) Expression of glutamate carboxypeptidase II in human brain. , Neurosci 144, 1361-1372.
- 23.M H Baslow. (2010) Evidence that the tri-cellular metabolism of N-acetylaspartate functions as the brain’s “operating system”: how NAA metabolism supports meaningful intercellular frequency-encoded communications. , Amino Acids 39, 1139-1145.
- 24.M H Baslow. (2008) The astrocyte surface NAAG receptor and NAAG peptidase signaling complex as a therapeutic target. , Drug News and Perspectives 21(5), 251-257.
- 25.D L Taylor, Davies S E C, T P Obrenovitch, Urenjak J, D A Richards et al. (1994) Extracellular N-acetylaspartate in the rat brain: In vivo determination of basal levels and changes evoked by high K+. , J. Neurochem 62, 2349-2355.
- 26.T N Sager, Fink-Jensen A, A J Hansen. (1997) Transient elevation of interstitial N-acetylaspartate in reversible global brain ischemia. , J. Neurochem 68, 675-682.
- 27.A J Shah, R de la Flor, Atkins A, Stone-Murphy J, L A Dawson. (2008) Development and application of liquid chromatography/tandom mass spectrometric assay for measurement of N-acetylaspartate, N-acetylaspartylglutamate and glutamate in brain superfusates and tissue extracts. , J. Chromatogr. B 876, 153-158.
- 28.Zur Nieden R, J W Deitmer. (2006) The role of metabotropic glutamate receptors for the generation of calcium oscillations in rat hippocampal astrocytes in situ. , Cereb. Cortex 16, 676-687.
- 29.Niwa K, Haensel C, Elizabeth Ross M, Iadecola C. (2001) Cyclooxygenase-1 participates in selected vasodilator responses of the cerebral circulation. , Circ. Res 88, 600-608.
- 30.Toth P, Tarantini S, Davila A, Valcarcel-Ares Noa, Tucsek M et al. (2015) Purinergic glio-endothelial coupling during neuronal activity: role of P2Y1 receptors and eNOS in functional hyperemia in mouse somatosensory cortex. , Am. J. Physiol. Heart Circ. Physiol., 309: H 1837 - H 1845.
- 31.Kinoshita K, Arai K, Kawaura K, Hiyoshi T, Yamaguchi J-i. (2015) Development, validation, and application of a surrogate method for determining N-acetyl-l-aspartyl-l-glutamic acid levels in rat brain, plasma, and cerebrospinal fluid. , J. Chromatogr. B 1003, 1-11.
- 32.J A Filosa, H W Morrison, J A Iddings, Du W, Kim K J. (2016) Beyond neurovascular coupling, role of astrocytes in the regulation of vascular tone. , Neuroscience 323, 95-109.
- 33.Petit J-M, P J Magistretti. (2016) Regulation of neuron-astrocyte metabolic coupling across the sleep-wake cycle. , Neuroscience 323, 135-156.
- 34.Furukawi-Hibi Y, Nitta A, Fukumitsu H, Somiya H, Toriumi K et al. (2012) Absence of SHATI/Nat8l reduces social interaction in mice. , Neurosci. Let 526, 79-84.
- 35.M H Baslow. (2009) The languages of neurons: An analysis of coding mechanisms by which neurons communicate, learn and store information. , Entropy 11, 782-797.
- 36.M H Baslow. (2006) NAAG peptidase as a therapeutic target: Potential for regulating the link between glucose metabolism and cognition. Drug News and Perspectives. 19(3), 145-150.
Cited by (5)
- 1.Baslow Morris H, 2017, Rescuing Canavan disease: engineering the wrong cell at the right time, Journal of Inherited Metabolic Disease, 40(5), 627, 10.1007/s10545-017-0038-2
- 2.Manzhurtsev Andrei, Menschchikov Petr, Yakovlev Alexei, Ublinskiy Maxim, Bozhko Olga, et al, 2021, 3T MEGA-PRESS study of N-acetyl aspartyl glutamate and N-acetyl aspartate in activated visual cortex, Magnetic Resonance Materials in Physics, Biology and Medicine, 34(4), 555, 10.1007/s10334-021-00912-5
- 3.Menshchikov Petr, Ivantsova Anna, Manzhurtsev Andrei, Ublinskiy Maxim, Yakovlev Alexey, et al, 2020, Separate N‐acetyl aspartyl glutamate, N‐acetyl aspartate, aspartate, and glutamate quantification after pediatric mild traumatic brain injury in the acute phase, Magnetic Resonance in Medicine, 84(6), 2918, 10.1002/mrm.28332
- 4.Grønbæk-Thygesen Martin, Hartmann-Petersen Rasmus, 2024, Cellular and molecular mechanisms of aspartoacylase and its role in Canavan disease, Cell & Bioscience, 14(1), 10.1186/s13578-024-01224-6
- 5.Baslow Morris H., 2016, Is the tri-cellular N-acetylaspartylglutamate (NAAG) cycle related to the etiology of schizophrenia?, Schizophrenia Research, 178(1-3), 112, 10.1016/j.schres.2016.09.009