
Liquid Chromatography Tandem Mass Spectrometry Method for Determination of Febuxostat in Human Plasma to Support A Bioequivalence Study
Abstract
A reliable, selective and sensitive liquid chromatography-tandem mass spectrometry (LC-ESI-MS/MS) assay has been proposed for the determination of febuxostat in human plasma using indomethacin as the internal standard (IS). The analyte and IS were extracted from 200 µL of human plasma via liquid-liquid extraction using methyl tert-butyl ether. Chromatography was performed on Hypurity C18 (100 mm × 4.6 mm, 5 µm) column under isocratic conditions. Detection of analyte and IS was done by tandem mass spectrometry, operating in negative ionization and multiple reaction monitoring mode. The deprotonated precursor to product ion transitions monitored for febuxostat and indomethacin were m/z 315.1 →271.0 and 356.1→312.0 respectively. The limit of detection (LOD) and limit of quantitation (LOQ) of the method were 0.0025 µg/mL and 0.05 µg/mL respectively. The linear dynamic range validated for febuxostat was 0.05-6.00 µg/mL. The intra-batch and inter-batch precision (% CV) was ≤ 7.1 % while the mean extraction recovery was > 87 % for febuxostat across quality control levels. The method was successfully applied to a bioequivalence study of 80 mg febuxostat tablet formulation in 14 healthy Indian male subjects under fasting and fed condition. The reproducibility in the measurement of study data was demonstrated by reanalysis of 110 incurred samples.
Author Contributions
Academic Editor: Rajiv Kumar, Northeastern University & DFCI Harvard Medical School
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2013 Dinesh S. Patel, et al.
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Introduction
Febuxostat FEB2 is a non-purine urate lowering drug, used in the treatment and management of hyperuricemia and gout 1, 2, 3. Gout is a common disorder characterized by hyperuricemia and by acute and chronic consequences of urate crystal deposition in the joints and periarticular tissues 4 FEB is a selective inhibitor of both the oxidised and reduced forms of xanthine oxidase, the enzyme that catalyzes the synthesis of uric acid from hypoxanthine and xanthine4. It was approved by the US Food and Drug Administration for the management of hyperuricemia in adults with gout in February 2009 5. FEB is well absorbed from the gastrointestinal tract after oral administration with an oral bioavailability of 84 % and the time to reach peak plasma concentration is about 1 h in healthy volunteers 2, 6. The half life of FEB is 5-8 h and the volume of distribution ranges from 29 to 751 L/kg at steady state for oral dose of 10-300 mg. It is highly protein bound (99 %), primarily to albumin and gets extensively metabolized via phase II (glucuronidation) and phase I (oxidation) in the liver followed by excretion of metabolites in the urine and faeces 1, 7, 8.
FEB has been determined in bulk and pharmaceutical dosage forms by reversed-phase HPLC 9, 10, 11. However, few methods are reported for the determination of FEB in biological matrices 8, 12, 13, 14, which are mainly based on HPLC. Khosravan et al. 8 and Grabowski et al. 12 have estimated the plasma concentration of FEB by first precipitating the proteins with acetonitrile from 0.5 mL human plasma, followed by HPLC-fluorescence analysis. For both these methods the calibration curves was linear from 0.01-20 µg/mL and were applied to study the effect of food or antacid 8 and hydrochlorothiazide 12 on the pharmaco- kinetics and pharmacodynamics of FEB in healthy volunteers. A similar method employing protein precipitation has been proposed which had a sensitivity of 0.10 µg/mL 13. It was used to evaluate the pharmacokinetics/bioequivalence of 80 mg FEB tablets in healthy Indian subjects. Recently, Lukram et al. 14 have developed a UPLC-MS/MS method to determine FEB in human plasma with a lower limit of quantitation of 0.075 µg/mL. A study of impurity carryover and impurity profile in FEB drug substance has been presented by LC-MS/MS 15.
Thus, in the present study a reliable, selective and rapid validated LC-MS/MS method has been proposed for the determination of FEB in human plasma. The method requires only 200 µL human plasma for liquid-liquid extraction and demonstrates excellent performance in terms of ruggedness and efficiency (5.0 min per sample). The dynamic linear range was validated from 0.05 – 6.00 µg/mL. The method is selective in presence of four non-steroidal anti-inflammatory drugs (NSAID) namely ibuprofen, naproxen, diclofenac and aspirin and some commonly used medications by subjects. Ion suppression/enhancement was studied by post column infusion of analyte. The proposed method has been successfully applied to a bioequivalence study of 80 mg febuxostat tablet formulation in 14 healthy Indian males under fasting and fed condition. The reproducibility in the measurement of study data is demonstrated by incurred sample reanalysis.
Experimental
Chemicals and Materials
Reference standard material of febuxostat (100.4 %) and indomethacin (IS, 99.8 %) were procured from Zydus Cadila (Ahmedabad, India) and Fabrica Italiana Sintetici (Montecchio Maggiore (VI), Italy) respectively. HPLC grade methanol, analytical grade reagent ortho phosphoric acid (88.0 %), glacial acetic acid (99.8 %) and ammonium acetate (98.0 %) were obtained from S.D. Fine Chemicals Ltd. (Mumbai, India). HPLC grade methyl tert-butyl ether (MTBE) was procured from Mallinckrodt Baker Inc. (Phillipsburg, New Jersey, USA). Deionized water used for LC-MS/MS was prepared using Milli Q water purification system from Millipore (Bangalore, India). Control buffered (K3EDTA) human plasma was procured from Clinical Department, Cliantha Research India Limited (Ahmedabad, India) and was stored at –20 °C.
LC-MS/MS Instrumentation and Settings
A Shimadzu liquid chromatography system (Kyoto, Japan) with Hypurity C18 (100 mm × 4.6 mm, 5.0 µm) column from Thermo Scientific (NJ, USA) was used in the study. The mobile phase consisted of methanol: 10 mM ammonium acetate: glacial acetic acid (70:30:0.01, v/v/v). The auto sampler temperature was maintained at 4 °C and the flow rate was set at 0.80 mL/min for isocratic conditions. The total eluent from the column was split in 20:80 ratio; flow directed to the ISP interface was equivalent to 160 μL/min. Ionization and detection of analyte and IS was performed on a triple quadrupole mass spectrometer, API-3000 equipped with Turbo Ion spray®, from MDS SCIEX (Toronto, Canada) operating in the negative ion mode. Quantitation was done using MRM mode to monitor deprotonated precursor → product ion transition of m/z 315.1 → 271.0 for FEB and 356.1 → 312.0 for IS. All the parameters of LC and MS were controlled by Analyst software version 1.4.2. For FEB and IS the source dependant parameters maintained were Gas 1(Nebulizer gas): 11 psi, ion spray voltage (ISV): -4500 V, turbo heater temperature (TEM): 500 °C, entrance potential (EP): -10V, collision activation dissociation (CAD): 5 psi, curtain gas (CUR): 12 psi. The compound dependent parameters like declustering potential (DP), focusing potential (FP), collision energy (CE) and cell exit potential (CXP) were optimized at -30, -200, -20 and -11 V for FEB and -11, -180, -11 and -10 V for IS respectively. Quadrupole 1 and 3 were maintained at unit resolution, while the dwell set was at 800 and 400 ms for FEB and IS respectively.
Preparation of Standard Stock and Plasma Samples
The FEB standard stock solution of 1000 µg/mL was prepared by dissolving requisite amount in methanol. This was further diluted in the same diluent to get an intermediate solution of 120µg/mL concentration. The IS stock solutions of 100 µg/mL was prepared by dissolving requisite amount of indomethacin in methanol. IS working solution (3.00 µg/mL) was prepared using the stock solution in deionized water. All the above solutions were stored at 4 °C until use. The calibration standards (CS) at 0.05, 0.10, 0.20, 0.50, 1.00, 2.00, 4.00, 5.00 and 6.00 µg/mL and quality control (QC) samples (LLOQ QC (0.05 µg/mL), lower limit of quantitation quality control; LQC (0.15 µg/mL), low quality control; MQC-2 (0.80 µg/mL) & MQC-1 (1.80 µg/mL), medium quality control; HQC (4.50 µg/mL), high quality control; ULOQ QC (6.00 µg/mL), upper limit of quantitation quality control) were prepared by spiking blank plasma with respective working solutions (5 % of total volume of plasma). The spiked plasma samples at all the levels were stored at -20 °C for validation and subject sample analysis.
Protocol for Sample Preparation
Prior to analysis, spiked plasma samples were thawed in water bath and allowed to equilibrate at room temperature. The samples were adequately vortexed using a vortexer before pipetting. Aliquots of 200 µL plasma solution containing 10 µL of working solution of FEB and 190 µL blank plasma were transferred into screw tubes, 10 µL of deionized water (for CS and QC samples)/10 µL of methanol (for study samples), 100 µL working solution of IS (3.00 µg/mL) was added and vortexed to mix. To the same screw tubes, 50 µL of ortho- phosphoric acid solution (2.5 %, v/v) was added and vortexed again. Thereafter, 4.0 mL of MTBE was added, the tubes were caped and vortexed for 3 min. The samples were then centrifuged for 5 min at 1811 × g. The organic layer was transferred to correspondingly labeled tubes after freezing the aqueous layer in dry ice bath and the organic layer was evaporated to dryness under gentle stream of nitrogen (15 psi) at 40°C. The residue was taken up in 400 µL of reconstitution solution and 5.0 µL was used for injection in LC-MS/MS, in partial loop mode.
Methodology for Validation
The validation of the method was done following the USFDA guidelines 16. Test for selectivity was carried out in 12 different lots of blank human plasma including haemolysed & lipemic plasma collected with K3EDTA as an anticoagulant. From each of these 12 different lots, two replicates each of 190 µL were spiked with 10 µL of methanol. In the first set, the double blank plasma (without analyte and IS) was directly injected after extraction, while the other set was spiked with only IS before extraction. Further, one system suitability sample (SSS) at CS-2 (0.10 µg/mL) concentration and two replicates of LLOQ concentration (CS-1) were prepared by spiking 190 µL blank human plasma with 10 µL of respective working solutions of FEB.
The potential interferences from the common drugs were determined. These included acetaminophen, caffeine, cetrizine, chlorpheniramine maleate and pseudoephedrine, which are commonly used by human volunteers. Additionally, four NSAIDs drugs namely ibuprofen, naproxen, diclofenac and aspirin were studied for ionization (ion suppression/enhancement), analytical recovery (precision and accuracy) and chromatographic interference (interference with MRM of analyte and IS). Their stock solutions (100 µg/mL) were prepared by dissolving requisite amount in methanol. Further, working solutions (20.0 µg/mL) were prepared in methanol, spiked in plasma and analyzed under the same conditions at LQC and HQC levels in triplicate. These sets were processed along with freshly prepared calibration curve standards (CS) and two sets (8 samples) of qualifying QC samples (HQC, MQC-1, MQC-2 and LQC). As per the acceptance criteria, the % accuracy should be within 85 to 115 %. The MRM transitions in the negative ionization mode for ibuprofen (205/161), naproxen (229/185), diclofenac (294/250) and aspirin (179/137) were studied.
The design of the carry over experiment involved the following sequence of injections i.e. double blank plasma sample à two samples of LLOQ à double blank plasma à ULOQ sample à double blank plasma to check for any interference due to carry over. The linearity of the method was determined by analysis of six calibration curves containing nine non-zero concentrations. The area ratio response for FEB/IS obtained from multiple reaction monitoring was used for regression analysis. Each calibration curve was analyzed individually by using least square weighted (1/x2) linear regression. The lowest standard on the calibration curve was accepted as the LLOQ, if the analyte response was at least ten times more than that of drug free (blank) extracted plasma.
Intra-batch and inter-batch (on three consecutive validation days) accuracy and precision were evaluated at six QC levels (LLOQ QC, LQC, MQC-2, MQC-1, HQC and ULOQ QC) in six replicates for FEB. The deviation (% CV) at each concentration level from the nominal concentration was expected to be within ±15 %. Similarly, the mean accuracy should not vary by ±15 % except for the LLOQ where it can be ±20 % of the nominal concentration. Also, 2/3 quality control samples should meet the criteria of ±15 % of nominal concentration. Reinjection reproducibility was performed by re-injecting one entire validation batch.
The relative recovery, matrix effect and process efficiency were evaluated as reported previously 17 at HQC, MQC-1, MQC-2 and LQC levels in six replicates. Relative recovery (RE) was calculated by comparing the mean area response of samples spiked before extraction to that of samples spiked after extraction at each QC level. The recovery of IS was similarly estimated. Absolute matrix effect (ME) was assessed by comparing the mean area response of samples spiked after extraction with the mean area response of neat standard solutions prepared in reconstitution solution. The overall ‘process efficiency’ (% PE) was calculated as (ME × RE)/100. The relative matrix effect on analyte quantification was also checked in eight different batches/lots of K3EDTA plasma including haemolysed and lipemic plasma. From each batch, four samples at LQC and HQC levels was prepared (spiked after extraction) and checked for the % accuracy and precision (% CV). The deviation of the standards and QCs should not be more than ±15 %.
Matrix ion suppression effects on the MRM LC-MS/MS sensitivity were evaluated by the post column analyte infusion experiment 18.A standard solution containing 1.80 µg/mLof FEB and 3.00 µg/mLof IS in mobile phase was infused post column via a ‘T’ connector into the mobile phase at 5.0 µL/min employing Harvard infusion pump. Aliquots of 5.0 µL of extracted 6.00 µg/mLsample and double blank plasma sample were then injected and MRM LC-MS/MS chromatograms were acquired for FEB and IS.
All stability results were evaluated by measuring the area ratio response (FEB/IS) of stability samples against freshly prepared comparison standards at LQC and HQC levels. Stock solutions of FEB and IS were checked for short term stability at room temperature and long term stability at 4 °C. The solutions were considered stable if the deviation from nominal value was within ±10.0 %. Bench top stability, processed sample stability at room temperature and at refrigerated temperature (4 °C), freeze thaw stability and long term stability at -20 °C were performed at LQC and HQC levels using six replicates at each level. To meet the acceptance criteria, the % CV and % accuracy should be within ±15 %.
Dilution integrity experiment was carried out at 5 times the ULOQ concentration i.e. 30.0 µg/mL and at HQC level for FEB. Six replicate samples each of 1/10 of 5 × ULOQ (3.00 µg/mL) and 1/10 of HQC (0.45 µg/mL) concentration were prepared and their concentrations were calculated, by applying the dilution factor of 10 against the freshly prepared calibration curve for FEB.
Bioequivalence Study and Incurred Sample Reanalysis
The bioequivalence study was “An open label, randomized, two period, two treatment, two sequence, balanced, crossover, single dose evaluation of relative oral bioequivalence of test (80 mg febuxostat tablets of a generic company) and a reference (ULORIC®, 80 mg febuxostat tablets from Takeda Pharmaceuticals America, Inc.,Deerfield, IL 60015, USA) formulation in 14 healthy Indian subjects under fasting and fed conditions”. All the subjects were informed of the aim and risk involved in the study and written consent were obtained. The inclusion criteria for volunteer selection was based on the age (18-45 years), body mass index (between 18.5 and 24.9 kg/height2), general physical examination, electrocardiogram and laboratory tests like hematology, blood chemistry, urine examination and immunological tests. The work was approved and subject to review by an Institutional Ethics Committee. The procedures followed while dealing with human subjects were based on International Conference on Harmonization, E6 Good Clinical Practice (ICH, E6 GCP) guidelines 19. The subjects for study were fasted 10 h before administration of the drug formulation. Further, under fed conditions the subjects were given high fat and high calorie breakfast (consisting of 200 mL milk with 16 gm sugar, 80 gm of black gram, two slices of bread with butter and two cheese cutlets, total 969 calories) 30 min prior to giving the drug under investigation. Blood samples were collected in vacutainers containing K3EDTA anticoagulant before (0.0 h) and at 0.33, 0.66, 1.0, 1.33, 1.66, 2.0, 2.33, 2.66, 3.0, 3.5, 4.0, 4.5, 5.0, 6.0, 8.0, 10.0, 12.0 and 24.0 h of administration of drug under fast and fed conditions. Blood samples were centrifuged at 2415 × g at 4 °C for 15 min and plasma was separated, stored at -20 °C until use. The pharmacokinetic parameters of FEB were estimated by non-compartmental model using WinNonlin software version 5.2.1 (Pharsight Corporation, Sunnyvale, CA, USA). An incurred sample re-analysis (ISR) was also conducted by computerized selection of 110 subject samples (10 % of total study samples analyzed) near Cmax and the elimination phase for both studies. The results obtained were compared with the data obtained earlier for the same sample using the same procedure. The percent change in the value should not be more than ±20 % 20.
Results and Discussion
Method Development
The tuning of MS parameters was carried out in negative ionization mode for FEB and IS as both the drugs have carboxylic acid group. The analyte and IS gave predominant singly charged deprotonated precursor M- ions at m/z of 315.1 and 356.1 for FEB and IS respectively in Q1 full scan spectra. In the product ion mass spectrum of FEB, the most abundant and consistent ion was observed at m/z 271.0, which can be attributed to the loss of CO2 from the precursor ion (Figure 1a) by applying -20 eV collision energy. The major product ion for IS in Q3 mass spectra was found at m/z 312.0 due to the elimination of CO2 from the precursor ion (Figure 1b). To attain an ideal Taylor cone and a better impact on spectral response, an optimum potential of 4500 V was kept which gave consistent and stable signal. Fine tuning of nebulizer gas and CAD gas was done to get a consistent and stable response.
Figure 1.Product ion mass spectra of (a) febuxostat (m/z 315.1 → 271.0 , scan range 40-400 amu) and (b) indomethacin (IS, m/z 356.1 → 312.0, scan range 50-400 amu) in negative ionization mode.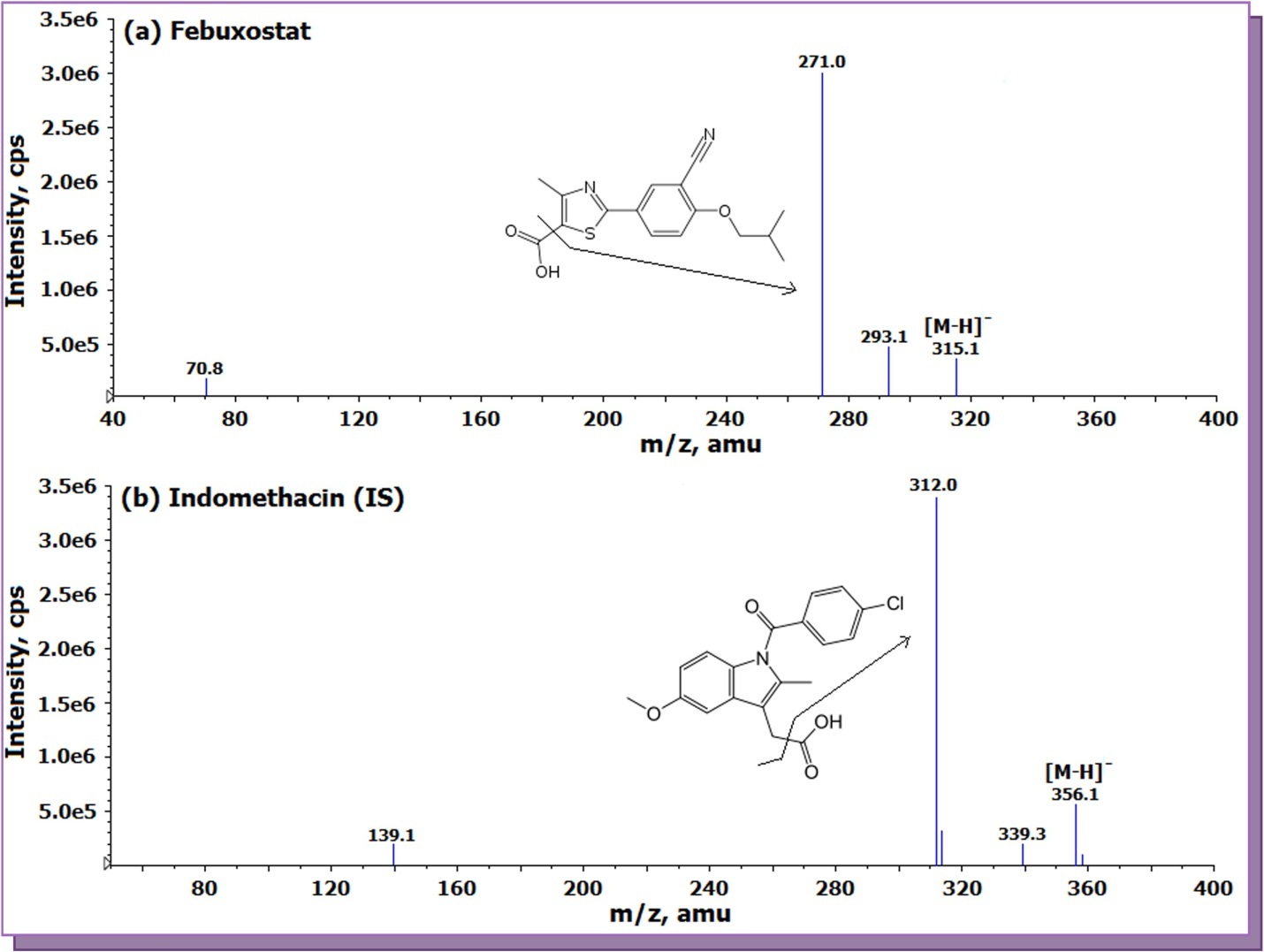
To develop a reliable and efficient chromatography, number of trials were conducted on three Thermo Scientific analytical columns namely BDS Hypersil C18 (100 mm × 4.6 mm, 5.0 µm), Hypurity C8 (100 mm × 4.6 mm, 5.0 µm), and Hypurity C18 (100 mm × 4.6 mm, 5.0 µm). Two previous methods 8, 12 have used Phenomenex Capcell Pak C18 column, however, the chromatography was not shown. Efforts were made to attain high sensitivity with good peak shapes, shorter run time, high throughput and to achieve desired selectivity with minimum matrix interference. Different volume ratios (50:50, 60:40, 70:30 and 80:20, v/v) of methanol-water and acetonitrile-water combinations were tried as mobile phase, along with glacial acetic acid and ammonium acetate (2-20 mM) on these columns. Further, the effect of flow rate was also studied from 0.3 to 1.0 mL/min, which was also responsible for acceptable chromatographic peak shapes. Efficient chromatography with adequate retention, response and peak shape was possible using a mobile phase consisting of methanol: 10 mM ammonium acetate: glacial acetic acid (70:30:0.01, v/v/v) at a flow rate of 0.8 mL/min on Hypurity C18 column. The response on all three columns was comparable; however, the run time was > 5 min on Hypurity C8 column. BDS Hypersil C18 offered consistent and adequate response, nevertheless the peak shapes and retention was more superior on Hypurity C18 for all QC samples and hence was selected in the present work. This could be due to higher carbon loading (13 %) in Hypurity C18 compared to BDS Hypersil C18 (11 %) and Hypurity C8 (8 %) columns. The retention time observed for FEB and IS was 2.48 and 3.73 in a run time of 5.0 min. The reproducibility of retention times for the analyte, expressed as %CV was ≤ 0.7 % for 100 injections on the same column. The maximum on-column loading (at ULOQ) of FEB per sample injection volume was 15 ng. Ideally, a deuterated analogue should be the first-choice internal standard, but due to its unavailability, a general IS was used to minimize analytical variation due to solvent evaporation, and ionization efficiency. Indomethacin, used as an IS in the present study had similar chromatographic behavior and was easily eluted along with the analyte. There was no effect of IS on analyte recovery, sensitivity or ion suppression.
All three previous reports 8, 12, 13 have employed protein precipitation (PP) with acetonitrile for quantitative extraction of FEB from human plasma. Though protein precipitation was tried initially (due to high protein binding of the drug) using methanol and acetonitrile as precipitating agents but the response was inconsistent with some ion suppression at LLOQ and LQC levels. Further, LLE was carried out in different solvent systems like diethyl ether, n-hexane and MTBE. The recovery obtained in all the three solvents was low (~50-60 %) and inconsistent. Thus, ortho- phosphoric acid was added to break the drug-plasma binding and to maintain the analyte and IS in their unionized state. Compared to other solvents, quantitative and consistent recovery (> 85 %) was obtained for FEB and IS all QCs levels in MTBE and was selected for the present work. In order to avoid excess blood loss especially during subject sample analysis, only 200 µL of human plasma was used for sample processing by liquid-liquid extraction, which provided adequate recovery for febuxostat. Although the recovery of febuxostat was almost 100 % in the UPLC-MS/MS 14, however, the plasma volume for FEB extraction in the present work is less compared to other methods reported in biological samples 8, 12, 13, 14.
System Suitability, Selectivity, Interference and Carryover Check
During method validation, the precision (%CV) of system suitability test was observed in the range of 0.14 to 0.45 % for the retention time and 1.2 to 2.1 % for the area response of FEB and IS. All plasma samples studied for selectivity check were found free from any endogenous interference. Figure 2 (a-c) demonstrates the selectivity results with the chromatograms of double blank plasma (without IS), blank plasma (with IS), peak response of FEB at LLOQ concentration. No interference was observed for commonly used medications like acetaminophen, caffeine, cetrizine, chlorpheniramine maleate, and pseudoephedrine, which is apparent from the real subject sample chromatograms of FEB at 0.75 h after oral administration of 80 mg tablet formulation (Figure 2d). Management of gout generally requires use of anti-inflammatory drugs along with urate-lowering agents 21. None of the NSAIDs drugs studied in the present work interfered in the determination of FEB. Under the optimized experimental conditions, the retention time for ibuprofen, naproxen, diclofenac and aspirin was observed at 2.81, 2.65, 3.13 and 2.20 min respectively. However, due to their different MRM transitions there was no interference in the quantification of FEB. The % accuracy results for FEB at both the QC levels was within 97.4 to 104.3 %. Carry-over evaluation was performed to ensure that it does not affect the accuracy and precision of the proposed method. Almost negligible area (less than 0.45 % of LLOQ area) was observed in double blank plasma run after ULOQ, which suggests no carry-over of the analyte in subsequent runs. Moreover, no ghost peaks appeared during the analysis of blank samples.
Linearity, Sensitivity, Accuracy and Precision
All six calibration curves were linear over the concentration range of 0.05 – 6.00 µg/mL with correlation coefficient r2 0.9989. A straight-line fit was made through the data points by least square regression analysis to give the mean linear equation y = (0.7626 ± 0.0378) x + (0.0040 ± 0.0035) where y is the peak area ratio of the analyte/IS and x the concentration of the analyte. The accuracy and precision (% CV) observed for the calibration curve standards ranged from 95.3 to 105.0 % and 1.7 to 5.3 % respectively. The lowest concentration (LLOQ) in the standard curve that can be measured with acceptable accuracy and precision was 0.05 µg/mL at a signal-to-noise ratio (S/N) ≥ 50 and a limit of detection (LOD) 0.0025 µg/mL. Based on the response it was possible to lower the LLOQ five folds, however it was not required based on subject sample analysis results. The sensitivity (0.05 µg/mL) achieved is higher compared to the work of Lukram et al. 14 (0.075 µg/mL) by UPLC-MS/MS.
The intra-batch and inter-batch precision and accuracy were established from validation runs performed at different QC levels (Table 1). The intra-batch precision (% CV) ranged from 0.9 to 2.8 and the accuracy was within 97.0 to 102.9 %. For the inter-batch experiments, the precision varied from 1.6 to 7.1 and the accuracy was within 100.0 to 105.6 %.
Table 1. Intra-batch and inter-batch accuracy and precision for febuxostat| QC | Conc. added(µg/mL) | Intra-batch | Inter-batch | ||||||
| n | Mean conc. found(µg/mL)a | Accuracy (%) | CV(%) | n | Mean conc. found(µg/mL)b | Accuracy (%) | CV(%) | ||
| LLOQ | 0.05 | 6 | 0.05 | 100.0 | 1.4 | 18 | 0.05 | 100.0 | 1.6 |
| LQC | 0.15 | 6 | 0.15 | 100.0 | 1.8 | 18 | 0.15 | 100.0 | 5.6 |
| MQC-2 | 0.80 | 6 | 0.82 | 102.5 | 0.9 | 18 | 0.83 | 103.8 | 2.7 |
| MQC-1 | 1.80 | 6 | 1.83 | 101.7 | 1.5 | 18 | 1.90 | 105.6 | 6.5 |
| HQC | 4.50 | 6 | 4.63 | 102.9 | 2.5 | 18 | 4.58 | 101.8 | 7.1 |
| ULOQ | 6.00 | 6 | 5.82 | 97.0 | 2.8 | 18 | 6.08 | 101.3 | 4.8 |
Recovery, Matrix Effect, Matrix Factor, Ion Suppression
The relative recovery, absolute matrix effect and process efficiency data for FEB is presented in Table 2. The relative recovery of the analyte is the ‘true recovery’, which is unaffected by the matrix as it is calculated by comparing the area ratio response (analyte/IS) of extracted (spiked before extraction) and unextracted (spiked after extraction) samples. The relative recovery obtained for FEB and IS was > 85 % at all QC levels. Further, the relative matrix effect which compares the precision (% CV) values between different lots (sources) of plasma (spiked after extraction) samples varied from 0.4 to 3.6 for FEB at the LQC and HQC levels. Qualitative assessment of ion suppression/enhancement was obtained through post column analyte infusion experiment for FEB, which is not available in reported methods 8, 12, 13, 14. Results of post-column analyte infusion experiment in Figure 3 indicate no ion suppression or enhancement at the retention time of FEB and IS. A minor suppression in the response was observed at 1.6 min, however, it did not interfere in the quantitation. The average matrix factor value calculated as the response of post spiked sample/response of neat solution (in reconstitution solution) at the LLQC level was 0.97, which indicates a minor suppression of about 3 %.
Table 2. Absolute matrix effect, relative recovery and process efficiency for febuxostat| A(%CV) | B(%CV) | C(%CV) | Absolute matrix effect(B/A × 100) | Relative recovery(C/B × 100) | Process efficiency(C/A × 100) |
| LQC | |||||
| 61958 (2.87) | 59170 (3.84) | 52484 (2.04) | 95.5 (99.4)a | 88.7 (85.1)a | 84.7 (84.6)a |
| MQC-2 | |||||
| 309740 (2.04) | 297970 (1.65) | 260723 (2.26) | 96.2 (96.8)a | 87.5 (86.2)a | 84.2 (83.4)a |
| MQC-1 | |||||
| 659972(1.38) | 638853(1.72) | 571135(0.93) | 96.8(97.5)a | 89.4 (87.6)a | 86.5(85.4)a |
| HQC | |||||
| 1551991 (3.30) | 1513192(2.75) | 1360359 (2.51) | 97.5 (97.4)a | 89.9 (89.1)a | 87.7 (86.8)a |
Figure 2.MRM ion-chromatograms of febuxostat (m/z 315.1 → 271.0 ) and indomethacin (IS, m/z 356.1 → 312.0) in (a) double blank plasma (without analyte and IS), (b) blank plasma with IS, (c) febuxostat at LLOQ and IS (d) real subject sample at 0.75 h after administration of 80 mg dose of febuxostat tablet.
Figure 3.Representative post column analyte infusion MRM LC-MS/MS overlaid chromatograms for febuxostat and indomethacin (a) Exact ion current (XIC) chromatogram of Febuxostat (m/z 315.1 → 271.0 ) (b) XIC of indomethacin (IS, m/z 356.1 → 312.0).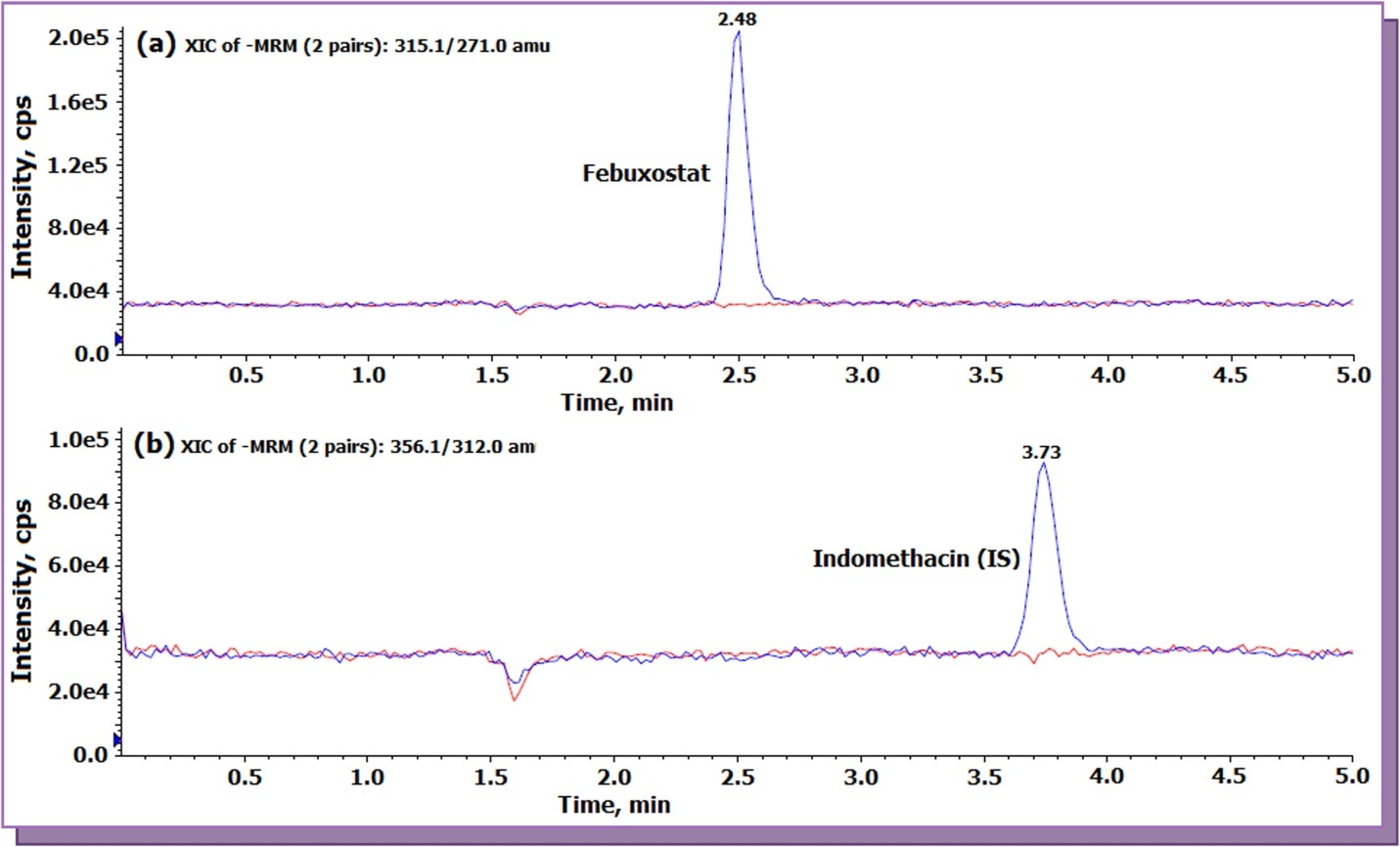
Stability and Dilution Reliability
Stability experiments were performed to evaluate the analyte stability in stocks solutions and in plasma samples under different conditions, simulating the same conditions which occurred during study sample analysis. The stock solution of FEB was stable at room temperature for 7 h and at 4 °C for 182 days. The intermediate stock solution of FEB in methanol was stable at 4 °C for 12 days with % change of 0.5. FEB was found stable in controlled plasma at room temperature up to 24 h and for six freeze and thaw cycles. The analyte in extracted plasma samples were stable for 96 h under refrigerated conditions (4 °C) and for 96 h under room temperature. The spiked plasma samples of FEB stored at -20 °C for long term stability were found stable for minimum period of 75 days. The values for the percent change for all the stability experiments are compiled in Table 3
Table 3. Stability experiments| Stability | Storage condition | Level | Mean stability sample a ±SD (µg/mL) | % change |
| Bench top stability | Room temperature (24h) | LQC | 0.15 ± 0.0019 | 0.0 |
| HQC | 4.69 ± 0.0426 | 4.2 | ||
| Processed sample stability | Auto sampler(4C, 96h) | LQC | 0.15 ± 0.0026 | 0.0 |
| HQC | 4.10 ± 0.0787 | -8.9 | ||
| Room temperature (96h) | LQC | 0.15 ± 0.0013 | 0.0 | |
| HQC | 4.12 ± 0.1389 | -8.4 | ||
| Freeze and thaw stability | After 6th cycle at- 20C | LQC | 0.15 ± 0.0006 | 0.0 |
| HQC | 4.26 ± 0.0380 | -5.3 | ||
| Long term stability | 75 days at- 20C | LQC | 0.14 ± 0.0022 | -6.7 |
| HQC | 4.23 ± 0.0450 | -6.0 |
The precision values for dilution integrity of 1/10 of 5 × ULOQ (3.00 µg/mL) and 1/10 of HQC (0.45 µg/mL) concentration were 2.0 and 1.5 %, while the accuracy results were 98.2 and 101.6%, respectively, which is within the acceptance limit of 15 % for precision (% CV) and 85 to 115 % for accuracy.
Application of the Method on Human Subjects and Incurred Sample Reanalysis
The validated method was applied for a bioequivalence study of FEB in 14 healthy Indian male subjects who received 80 mg test and reference formulations of FEB under fasting and fed condition. The mean pharmacokinetic profiles for the studies are presented in Figure 4. In all approximately 1682 samples including the calibration, QC and volunteer samples were run and analyzed successfully. The mean pharmacokinetic parameters obtained for the test and reference formulation for both the studies are presented in Table 4a. It has been shown that food causes a decrease in the rate and extent of absorption of FEB 8. The Cmax and t1/2 values obtained under fasting were higher compared to fed conditions, while the Tmax value under fed condition was about three times higher than that for fasting. All the pharmacokinetic parameters obtained under fasting were comparable with the work of Menon et al. 13 with healthy Indian volunteers. The mean log-transformed ratios of the parameters and their 90% CIs were all within the defined bioequivalence range (Table 4b). The % change in the randomly selected subject samples for incurred sample reanalysis was within 8.0 to – 9.7 % as shown in Figure 5 This authenticates the reproducibility and ruggedness of the proposed method.
Figure 4.Mean plasma concentration-time profile of febuxostat after oral administration of test (80 mg, febuxostat tablets from a generic company) and a reference (ULORIC®, 80 mg febuxostat tablets from Takeda Pharmaceuticals America, Inc., Deerfield, IL 60015, USA) formulation to 14 healthy Indian subjects under fasting and fed condition.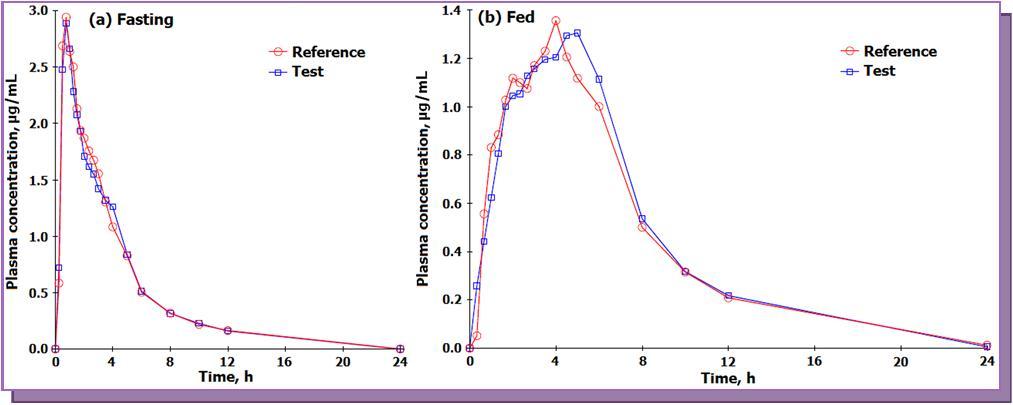
| Parameter | Fasting | Fed | ||
| Test | Reference | Test | Reference | |
| Mean ±SD | Mean ±SD | Mean ±SD | Mean ±SD | |
| Cmax (µg /mL) | 2.76+ 1.27 | 2.95+ 0.85 | 1.32+ 0.62 | 1.26+ 0.67 |
| Tmax (h) | 1.51 + 1.42 | 1.34 + 1.24 | 4.31 + 1.94 | 4.10 + 1.78 |
| t1/2 (h) | 3.42 + 0.96 | 3.56 + 1.06 | 2.78 + 0.68 | 3.00 + 0.94 |
| AUC0 - 24h (h.µg/ml) | 10.36 + 2.72 | 10.47 + 3.15 | 9.32 + 2.73 | 9.28 + 3.49 |
| AUC0-inf (h.µg/ml) | 11.09 + 2.89 | 11.35 + 3.36 | 10.07 + 3.03 | 9.92 + 3.56 |
| Kel (1/h) | 0.238 + 0.060 | 0.213 + 0.070 | 0.262 + 0.054 | 0.250 + 0.068 |
| Parameter | Ratio (test/ reference),% | 90% CI (Lower – Upper) | Power | Intra subject variation, % CV | ||||
| Fast | Fed | Fast | Fed | Fast | Fed | Fast | Fed | |
| Cmax(µg/mL) | 93.5 | 104.6 | 85.6 – 101.3 | 95.2 – 112.0 | 0.9940 | 0.9812 | 12.5 | 16.5 |
| AUC0-24 (h.µg/mL) | 99.7 | 102.3 | 91.6 – 108.5 | 94.8 – 110.3 | 0.9935 | 0.9978 | 12.6 | 11.2 |
| AUC0-inf (h.µg/mL) | 98.2 | 102.6 | 89.9 – 107.4 | 95.6 – 110.1 | 0.9902 | 0.9989 | 13.3 | 10.5 |
Figure 5.Graphical representation of results for 110 incurred samples for febuxostat.
Conclusion
Very few methods have been reported in literature for determination of FEB in biological matrices, with limited information on method development and validation. Thus, the objective of this work was to develop a selective, rugged and a high throughput method for the estimation of FEB in human plasma by LC-MS/MS. The liquid-liquid extraction procedure employed in the present work gave consistent and quantitative recoveries for FEB at all QC levels. The current method is more sensitive compared to all other methods reported in human plasma. The maximum on-column loading at ULOQ was 15 ng for 5.0 µL injection volume,which helps in maintaining the efficiency and the lifetime of the column. Moreover, the limit of quantification is low enough to monitor at least five half-lives of FEB concentration with good intra and inter-assay reproducibility (% CV) for the quality controls. The method was selective in presence four NSAIDs and some commonly used medications by human volunteers. The sensitivity of the proposed method is adequate to support a wide range of pharmacokinetic/bioequivalence studies. The study under fast and fed conditions proves that although food causes a decrease in the rate and extent of absorption of febuxostat, it does not lead to clinically significant change in febuxostat pharmacokinetics in healthy Indian volunteers. Thus febuxostat can be administered with or without food during anti-hyperuricemic therapy for the treatment of gout. The reproducibility in the measurement of subject samples is efficiently demonstrated by incurred sample reanalysis, which is not reported in earlier methods.
Acknowledgements
The authors are indebted to Mr. Vijay Patel, Executive Director, Cliantha Research Ltd., Ahmedabad for providing necessary facilities to carry out this work. We gratefully acknowledge Mr. Anshul Dogra, Study Director and Mrs. Arpana Prasad, R & D Supervisor, Cliantha Research India Ltd. for their continuous support, motivation and assistance during the course of this project.
References
- 4.M A Becker, H R Schumacher, L R Espinoza, A F Wells, P A MacDonald. (2010) . , Arthritis Res. Ther. 12, R 63.
- 8.Khosravan R, Grabowski B, J T Wu, Joseph-Ridge N, Vernillet L. (2007) . , Br. J. Clin. Pharmacol 65, 355-363.
- 12.Grabowski B, Khosravan R, J T Wu, Vernillet L, Lademacher C. (2010) . , Br. J. Clin. Pharmacol 70, 57-64.
- 15.M H Kadivar, P K Sinha, Kushwah D, Jana P, Sharma H. (2011) . , J. Pharm. Biomed. Anal 56, 749-757.
- 16.May2001Guidance for Industry, Bionanlytical Method Validation, US Department of Health and Human Services, Food and Drug Administration Centre for Drug Evaluation and Research (CDER), Centre for Veterinary Medicine (CVM).
- 18.King R, Bonfiglio R, Fernandez-Metzler C, Miller-Stein C, Olah T. (2000) . , J. Am. Soc. Mass Spectrom 11, 942-950.
Cited by (7)
This article has been cited by 7 scholarly works according to:
Citing Articles:
Journal of Chromatography B (2024) Crossref
U. Thorsteinsdóttir, H. Runolfsdottir, F. Eiríksson, I. M. S. Agustsdottir, V. Edvardsson et al. - Journal of chromatography. B, Analytical technologies in the biomedical and life sciences (2024) Semantic Scholar
Melchor Alpízar, José de Jesús Reséndiz, Elisa García Martínez, Sanyog Dwivedi, Miguel Alejandro Trejo - Pharmaceutics (2024) Semantic Scholar
Pharmaceutics (2024) Crossref
G. Pabani, R. Mehta, P. Shah - Asian Journal of Clinical Pediatrics and Neonatology (2020) Semantic Scholar
Microchemical Journal (2019) Crossref
Noha M. Hosny, N. N. Atia, S. El-Gizawy - Microchemical journal (Print) (2019) Semantic Scholar
Kareem M. Younes, Ehab F El-Kady, E. Elzanfaly - Journal of Chromatographic Science (2016) Semantic Scholar
Journal of Chromatographic Science (2016) Crossref
International Journal of Electrochemical Science (2016) Crossref
P. G. Sunitha, K. Ilango - (2014) Semantic Scholar
European Journal of Chemistry (2014) Crossref