
Abstract
Fungal infections increased substantially in the last years, becoming a relevant public health problem. Many of these infections account for high rates of morbidity and mortality. The emergence of resistant fungal clinical isolates have also motivate studies to find new antifungal therapies. Candida albicans is an oportunistic pathogen and affects a great number of immunocompromised patients worldwide. The marine ecosystem has been considered a rich source of bioactive metabolites due to the complexity and originality of its structures. Proteins and peptides from marine organisms have been shown to have antiviral, anti-inflammatory, antimalarial, anticancer, antimicrobial and antifungal properties. Arenicins are antimicrobial peptides isolated from the marine lugworm Arenicola marina with 21 amino acid residues in a β-hairpin structure. Dihydrofolate reductase, exo-b-(1,3)-glucanase and sterol 14α-demethylase are essential C. albincas enzymes that take part in DNA, cell wall and membrane metabolism, respectively. The present study evaluates the interaction of arenicin with important enzymes of C. albicans related to cell wall, ergosterol and DNA metabolism in order to elucidate possible molecular targets. We showed through an in silico approach, that a single compound from a marine worm (A. marina), can bind to three C. albicans essential proteins. The interaction occurs in regions inside the active site or at least near, with amino acid residues evaluated as hot spots. Arenicin is a new promising antifugal drug. The next step is to investigate protein-protein interactions performed by DHFR, EBG and CYP51 and assess whether arenicin is able to disrupt essential interaction or not.
Author Contributions
Academic Editor: Cesar Amaral, DNA Diagnostic Laboratory, University of the State of Rio de Janeiro, Brazil
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2019 Lívia do Carmo Silva, et al.
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Introduction
Fungal infections increased substantially in the last years, becoming a relevant public health problem. Many of these infections account for high rates of morbidity and mortality. Candidiasis, an opportunistic fungal infection is caused by fungi belonging to the Candida genus 1. Although the antifungal agents used in clinical treatments of mycosis have sometimes proved efficient, they are restricted to few targets in fungal cells with high toxicity to humans 2. In addition, the problem increases with the emergence of resistant clinical isolates 3, 4, which makes the search for new antifungal therapies extremely relevant.
Since the discovery of spongouridine, molecular model of the first antiviral used as a therapeutic resource for Acquired Immunodeficiency Syndrome (AIDS), the biodiversity of the marine ecosystem has been considered a rich source of bioactive metabolites due to the complexity and originality of its structures 5. In this scenario, peptides from marine organisms have been shown to have antiviral, anti-inflammatory, antimalarial, anticancer, antimicrobial and antifungal properties 6, 7, 8.
Arenicins are antimicrobial peptides isolated from the marine lugworm Arenicola marina with 21 amino acid residues in a β-hairpin structure Figure 1. In vitro and in vivo studies have shown that arenicins exhibit a very potent bactericidal and antifungal effect against multi-resistant gram-negative bacteria and Candida albicans, respectively 9. In bacterial, Arenicin interacts with membrane lipids promoting membrane permeabilization and detachment besides affecting cytoplasm metabolism 10. However, the interaction of arenicin with important antifungal targets from Candida albicans is unknown.
Figure 1.The arenicin structure. The conformational structure of the peptide folds into an antiparallel beta-sheet comprising two strands, which are important for its antimicrobial activity. Arenicin has a high affinity for lipids and disrupt fungal and other microorganisms membrane.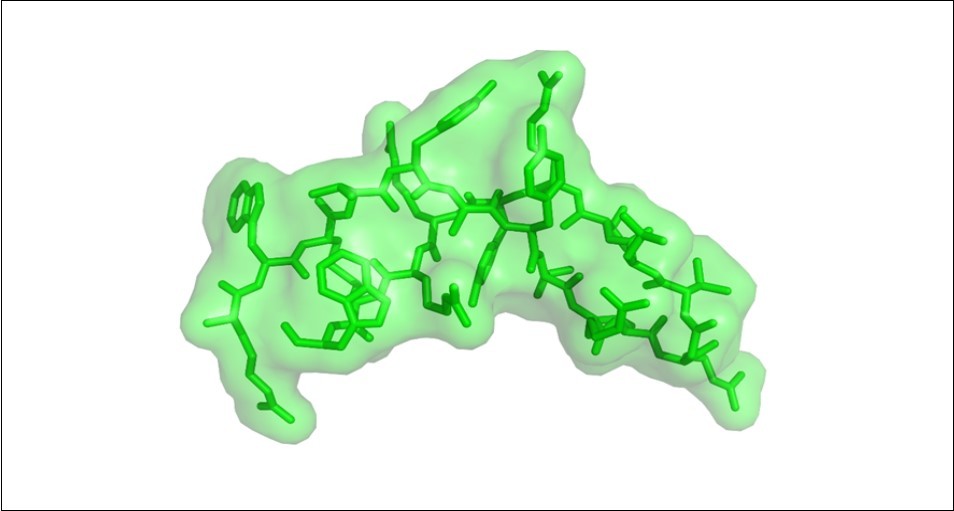
Dihydrofolate reductase (DHFR) catalyzes the reduction of dihydrofolic acid to tetrahydrofolic acid 11. The enzyme acts on the synthesis of purines, thymidylic acid, amino acids and the regulation of tetrahydrofolate levels in the cell, participating in proliferation and growth 12. The protein exo-b-(1,3)-glucanase (EBG) takes part in cell expansion and differentiation through cell wall remodeling. EBG is also secreted during pathogenesis 13. Finally, CYP51 (sterol 14α-demethylase) is a major drug target in the cytochrome P450 superfamily. This enzyme is related to ergosterol and membranes metabolism 14. All the enzymes described above are important to survivability and pathogenesis of Candida species. The identification of novel therapeutics can drive a more efficient treatment of immunocompromised patients that are susceptible to fungal diseases.
Molecular anchoring is an important computational tool in drug design and targeting. The purpose of the ligand-protein anchor is to predict the best interaction orientation of a linker with a protein of known three-dimensional structure 15. Thus, in this study we verified the interaction of arenicin with important enzymes of C. albicans related to cell wall, ergosterol and DNA metabolism in order to elucidate possible molecular targets.
Material and Methods
All the 3-D structures used in the analysis are available in the PDB (protein databank; https://www.rcsb.org/). KBDOCK server were used in order to assess protein domains and possible interaction between protein domains 16. The protein docking was performed by ClusPro 17. We used PyMol (https://pymol.org) for the visualization of the interface of interaction, the visualization of hot spots and for creating the figures presented in this manuscript .The hot spots in the proteins under study were identified by KFC2. The server offers an automated analysis of a protein complex interface. The server analyses the structural environment around amino acid residues and checks for already known hot spots environments determined experimentally. The hot spot prediction is based on characteristics regarding conformation specificity (K-FADE) and biochemical features such as hydrophobicity (K-CON) 18, 19.
Results and Discussion
The Arenicin Properties are Defined by its Amino Acid Composition
Arginine and valine account for 54% of the amino acid residues present in the arenicin structure Figure 2 and they account for the properties presented by the peptide. The large amount of arginine residues has a role in maintaining the conformational state of the peptide and its overall charge 20, 21, 22. Moreover, arginine residues may interact with active sites of proteins 23, 24, 25 that bind to phosphorylated substrates through hydrogen bonds. The large amount of valine residues helps the peptide folds into its β-hairpin structure Figure 326 and this is related to the presence of two non-hydrogen substituent attached to the valine C-beta carbon. In addition, valine plays a role in substrate recognition of hydrophobic ligands such as lipids 27, 28, which could explain the high affinity the peptide has for membranes.
Figure 2.The arenicin arginine and valine residues. Arginine and valine amino acids correspond to 54% of residues in the arenicin structure. They contribute to the conformational state of the peptide, the overall charge and affinity for ligands. Red – arginine; blue – valine; green – other amino acid residues.
Figure 3.Dihydrofolate reductase (DHFR) interaction with arenicin. DHFR is a homodimeric protein and the inhibitor marine compound arenicin binds to a loop within the active site of the protein. Arenicin anchors inside the active site and alter slightly the conformational structure of DHFR. Green – DHFR; red – arenicin.
Arenicin Binds to Residues in the Active Site of Dihydrofolate Reductase
The main feature of the conformational structure of DHFR is a central region comprised by beta-pleated sheets 29. The active site is surrounded by a domain that contain loops and regulate the interaction pattern with partner proteins and compounds 30. An efficient strategy for inhibiting DHFR from C. albicans is to target essential amino acid residues at the active site of the enzyme 31, 32, 33. Arenicin interacts with a loop inside the active site of DHFR Figure 4. There are five main amino acid residues (Ile-Pro-Gln-Lys-Phe) that contribute considerably for the stability of the interaction between DHFR and the arenicin peptide and they have been shown to link to other inhibitor compounds as well 33.
Figure 4.Arenicin anchors inside the active site of dihydrofolate reductase. The loop in the active site of DHFR (pink) contains the five amino acid residues Ile-Pro-Gln-Lys-Phe. The binding of the inhibitor compound arenicin within the active site of the enzyme might hinder the efficiency of interaction between DHFR and partner proteins and consequently reduces its activity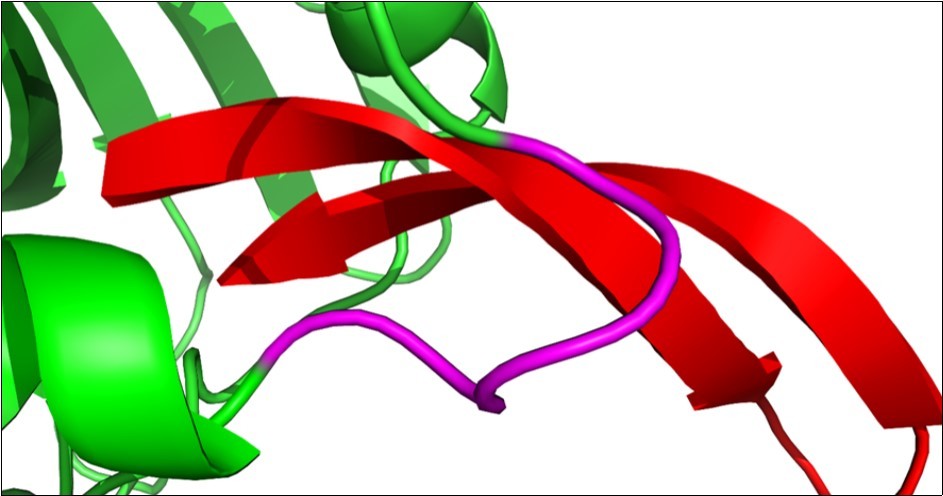
Arenicin Binds to Hot Spots Residues Near the Active Site of EBG
EBG folds into 8-barrel structure with both alfa and beta sheets, a typical conformation presented by the glycosyl hydrolase family. The active site is in a pocket within the protein structure 34. The pocket is energetically maintained by hydrogen-bonded interactions 35. Figure 5 shows the surface of EBG and the place where arenicin invades EBG towards the active site. We found seven hot spot residues on the EBG surface. They energetically contribute to the binding between the protein and the marine inhibitor. The main hot spot residues from EBG interacting with the inhibitor peptide are near residues belonging to the active site Figure 6. Arenicin bind to hot spot residues and changes the conformational structure of amino acids surrounding the active site of the protein, consequently inhibiting its activity.
Figure 5.Surface overview of EBG and arenicin interaction. Arenicin (red) binds to EBG and invades a pocket region of the protein that has amino acid residues belonging to the active site. Blue – EBG; red – arenicin; white – interface of interaction between the inhibitor peptide arenicin and EBG; green – hot spot residue.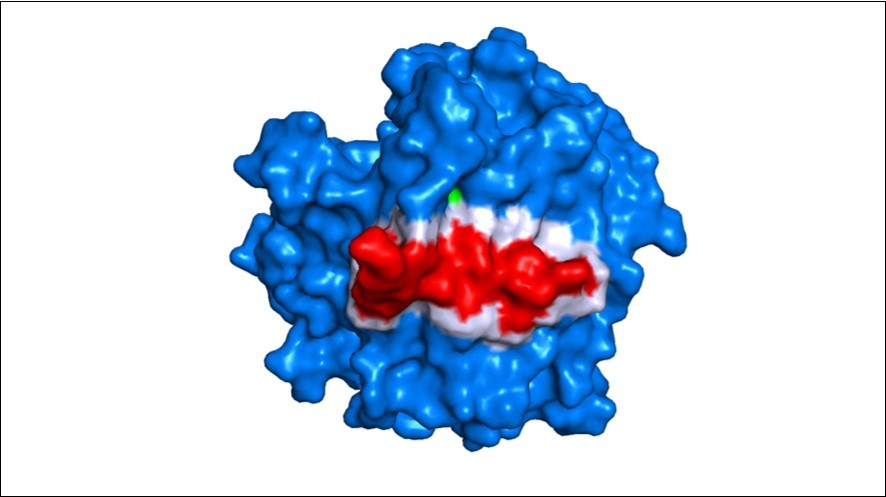
Figure 6.Arenicin and the pocket region of EBG. Arenicin (red) binds to EBG and invades a pocket region of the protein that has amino acid residues belonging to the active site (pink). Green amino acids are hot spots on EBG and the yellow amino acid residue was classified as a hot spot and belongs to the active site of EBG, an important residue for inhibiting assays.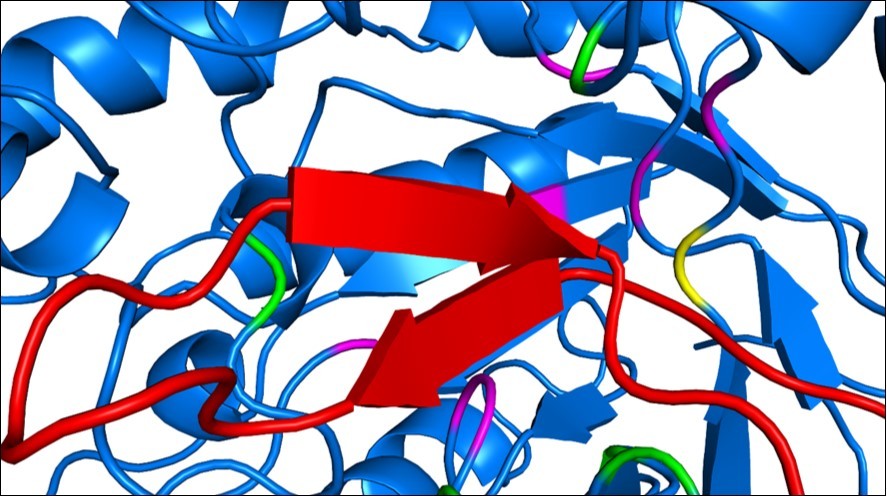
Arenicin is Surround by Hot Spots Residues in a Cleft of CYP51
CYP51 is required for ergosterol biosynthesis 36. It has been extensively investigated as the main target for azole antimicrobial drugs 37, 38, 39. We found 11 hot spot residues on the C. albicans CYP51 interface of interaction with the peptide arenicin Figure 7. The necessity of finding new target to fungal diseases is real, since azoles, although having the ability to inhibit CYP51, are toxic to mammals cells and lead to severe side effects in patients 40, 41, 42, 43. The marine peptide arenicin is held near the active site of the enzyme by the hydrophobicity in that area. Inhibition of CYP51 leads to disruption of the membrane and cell death. The marine peptide fits in a cleft of the protein, at the entrance of the active site, and is surrounded by hot spot residues, interacting with them via hydrogen bonds.
Figure 7.Arenicin surrounded by hot spot residues on CYP51 structure. Arenicin (red) binds to CYP51 (pink) and interacts with hot spot residues. Green amino acids are hot spots on CYP51, the conformation of the region of interaction changes, affecting the ability of the protein to perform its function.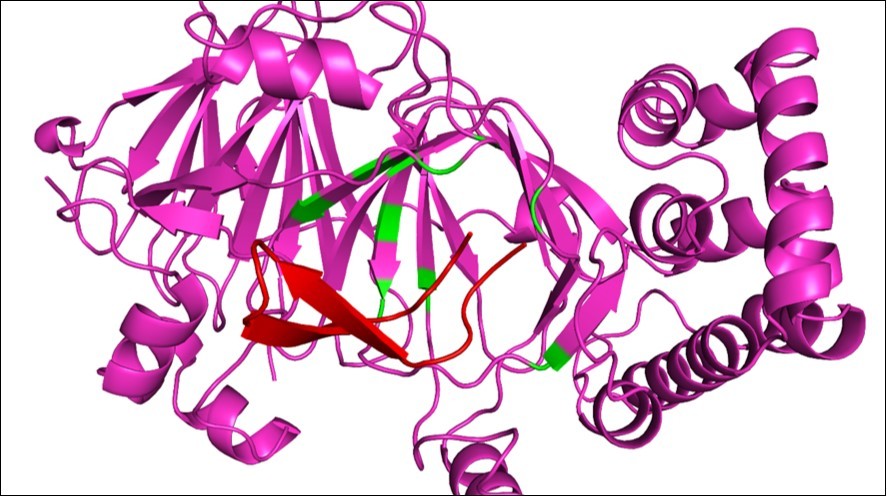
Concluding Remarks
The number of deaths of immunocompromised patients caused by opportunistic fungi, including C. albicans, has increased significantly 44. The therapeutics used against such infections are mostly based on azoles and other drugs that develop severe side effects in those patients. Here, we showed through an in silico approach, that a single compound from a marine worm (A. marina), can bind to three C. albicans essential proteins. The interaction occurs in regions inside the active site or at least near, with amino acid residues evaluated as hot spots. Arenicin is a new promising antifugal drug. The next step is to investigate protein-protein interactions performed by DHFR, EBG and CYP51 and assess whether arenicin is able to disrupt essential interaction or not.
References
- 1.Bongomin F, Gago S, Oladele R O, Denning D W.Global and Multi-National Prevalence of Fungal Diseases-Estimate Precision. de outubro de 2017;3(4) , J Fungi (Basel) 18.
- 2.Perfect J R.The antifungal pipeline: a reality check. Nat Rev Drug Discov. setembro de 2017;16(9):. 603-16.
- 3.RSN Brilhante, A Rodrigues PH de, de Alencar LP, Riello G B, Ribeiro J F et al.. Evidence of Fluconazole-Resistant Candida Species in Tortoises and Sea Turtles. Mycopathologia. dezembro de 2015-180.
- 4.Prigent G, Aït-Ammar N, Levesque E, Fekkar A, Costa J-M et al. (2017) Echinocandin Resistance in Candida Species Isolates from Liver Transplant Recipients. Antimicrob Agents Chemother.
- 5.Li T, Ding T, Li J. (2017) Medicinal Purposes: Bioactive Metabolites from Marine-derived Organisms. Mini Rev Med Chem. 26 de setembro de.
- 6.Lee Y, Phat C, Hong S-C.Structural diversity of marine cyclic peptides and their molecular mechanisms for anticancer, antibacterial, antifungal, and other clinical applications. Peptides. setembro de 2017;95:. 94-105.
- 7.Ruiz-Ruiz F, Mancera-Andrade E I, HMN Iqbal.Marine-Derived Bioactive Peptides for Biomedical Sectors: A Review. Protein Pept Lett. 24(2), 109-17.
- 8.Gogineni V, Hamann M T.Marine natural product peptides with therapeutic potential: Chemistry, biosynthesis, and pharmacology. , Biochim Biophys Acta Gen Subj. janeiro de 1862(1), 81-196.
- 9.Wang Y-H, Dong H-H, Zhao F, Wang J, Yan F et al.The synthesis and synergistic antifungal effects of chalcones against drug resistant Candida albicans. , Bioorg Med Chem Lett 26(13), 3098-102.
- 10.Andrä J, Jakovkin I, Grötzinger J, Hecht O, Krasnosdembskaya A D et al.Structure and mode of action of the antimicrobial peptide arenicin. , Biochem J. 15 de fevereiro de 410(1), 113-22.
- 11.Chen M J, Shimada T, Moulton A D, Harrison M, Nienhuis A W.Intronless human dihydrofolate reductase genes are derived from processed RNA molecules. Proc Natl Acad Sci USA. dezembro de 1982;79(23): 7435-9.
- 12.Urlaub G, Chasin L A.Isolation of Chinese hamster cell mutants deficient in dihydrofolate reductase activity. Proc Natl Acad Sci USA. julho de 1980;77(7): 4216-20.
- 13.Côté F, Hahn M G.Oligosaccharins: structures and signal transduction. Plant Mol Biol. dezembro de 1994;26(5):. 1379-411.
- 14.Marichal P, Gorrens J, Laurijssens L, Vermuyten K, C Van Hove et al.Accumulation of 3-ketosteroids induced by itraconazole in azole-resistant clinical Candida albicans isolates. Antimicrob Agents Chemother. novembro de 1999;43(11):. 2663-70.
- 15.Ferreira L G, Dos Santos RN, Oliva G, Andricopulo A D.Molecular docking and structure-based drug design strategies. , Molecules. 22 de julho de 20(7), 13384-421.
- 16.Ghoorah A W, Devignes M-D, Smaïl-Tabbone M, Ritchie D W.Classification and Exploration of 3D Protein Domain Interactions Using Kbdock. Methods Mol Biol. 2016, 91-105.
- 17.Kozakov D, Hall D R, Xia B, Porter K A, Padhorny D et al.The ClusPro web server for protein-protein docking. Nat Protoc. fevereiro de 2017;12(2):. 255-78.
- 18.Darnell S J, LeGault L, Mitchell J C.KFC Server: interactive forecasting of protein interaction hot spots. Nucleic Acids Res. 1ode julho de 2008;36(Web Server 265-269.
- 19.Darnell S J, Page D, Mitchell J C.An automated decision-tree approach to predicting protein interaction hot spots. Proteins. 1ode setembro de 68(4), 813-23.
- 20.Lipska A G, Sieradzan A K, Krupa P, Mozolewska M A, D’Auria S et al.Studies of conformational changes of an arginine-binding protein from Thermotoga maritima in the presence and absence of ligand via molecular dynamics simulations with the coarse-grained UNRES force field. , J Mol Model. março de 21(3), 64.
- 21.Vostrikov Soller KJ, Ha K N, Gopinath T, Veglia G.Effects of naturally occurring arginine 14 deletion on phospholamban conformational dynamics and membrane interactions. Biochim Biophys Acta. janeiro de 2015;1848(1 Pt B):. 315-22.
- 22.Wen L, Chen Y, Liao J, Zheng X, Yin Z.Preferential interactions between protein and arginine: effects of arginine on tertiary conformational and colloidal stability of protein solution. , Int J Pharm 478(2), 753-61.
- 23.Biswas A, Shukla A, RSK Vijayan, Jeyakanthan J, Sekar K.Crystal structures of an archaeal thymidylate kinase from Sulfolobus tokodaii provide insights into the role of a conserved active site Arginine residue. , J Struct Biol 197(3), 236-49.
- 24.Qin M, Song H, Dai X, Chen Y, Guo Z. (2018) Two active site arginines are critical determinants of substrate binding and catalysis in MenD, a thiamine-dependent enzyme in menaquinone biosynthesis. , Biochem J 19.
- 25.Fordwour O B, Wolthers K R.Active site arginine controls the stereochemistry of hydride transfer in cyclohexanone monooxygenase. Arch Biochem Biophys. 1ode outubro de 2018;659:. 47-56.
- 26.Kakuda K, Yamaguchi K-I, Kuwata K, Honda R. (2018) A valine-to-lysine substitution at position 210 induces structural conversion of prion protein into a β-sheet rich oligomer. Biochem Biophys Res Commun. 16 de outubro de.
- 27.Masbuchin A N, Rohman M S, Putri J F, Cahyaningtyas M. () Widodo null. 279(Val→Phe) Polymorphism of lipoprotein-associated phospholipase A(2) resulted in changes of folding kinetics and recognition to substrate. Comput Biol Chem. dezembro de 2015;59 Pt A:.
- 28.Moreau M, Takahashi H, Sari M-A, Boucher J-L, Sagami I et al.Importance of valine 567 in substrate recognition and oxidation by neuronal nitric oxide synthase. , J Inorg Biochem. julho de 98(7), 1200-9.
- 29.Matthews D A, Alden R A, Bolin J T, Freer S T, Hamlin R et al.Dihydrofolate reductase: x-ray structure of the binary complex with methotrexate. , Science 197(4302), 452-5.
- 30.Osborne M J, Schnell J, Benkovic S J, Dyson H J, Wright P E.Backbone dynamics in dihydrofolate reductase complexes: role of loop flexibility in the catalytic mechanism. , Biochemistry 40(33), 9846-59.
- 31.Liu J, Bolstad D B, Smith A E, Priestley N D, Wright D L et al.Structure-guided development of efficacious antifungal agents targeting Candida glabrata dihydrofolate reductase. Chem Biol. 22 de setembro de 2008;15(9):. 990-6.
- 32.Liu J, Bolstad D B, Smith A E, Priestley N D, Wright D L et al.Probing the active site of Candida glabrata dihydrofolate reductase with high resolution crystal structures and the synthesis of new inhibitors. Chem Biol Drug Des. janeiro de 2009;73(1):. 62-74.
- 33.Paulsen J L, Bendel S D, Anderson A C.Crystal structures of Candida albicans dihydrofolate reductase bound to propargyl-linked antifolates reveal the flexibility of active site loop residues critical for ligand potency and selectivity. Chem Biol Drug Des. outubro de 2011;78(4):. 505-12.
- 34.Cutfield S M, Davies G J, Murshudov G, Anderson B F, Moody P C et al.The structure of the exo-beta-(1,3)-glucanase from Candida albicans in native and bound forms: relationship between a pocket and groove in family 5 glycosyl hydrolases. , J Mol Biol 294(3), 771-83.
- 35.Sakon J, Adney W S, Himmel M E, Thomas S R, Karplus P A.. Crystal structure of thermostable family 5 endocellulase E1 from Acidothermus cellulolyticus in complex with cellotetraose. Biochemistry. 20 de agosto de 1996;35(33): 10648-60.
- 36.Kelly S L, Lamb D C, Jackson C J, Warrilow A G, Kelly D E.The biodiversity of microbial cytochromes P450. Adv Microb Physiol. 2003, 131-86.
- 37.AGS Warrilow, Martel C M, Parker J E, Melo N, Lamb D C et al.Azole binding properties of Candida albicans sterol 14-alpha demethylase (CaCYP51). Antimicrob Agents Chemother. outubro de 2010;54(10):. 4235-45.
- 38.Parker J E, Merkamm M, Manning N J, Pompon D, Kelly S L et al.Differential azole antifungal efficacies contrasted using a Saccharomyces cerevisiae strain humanized for sterol 14 alpha-demethylase at the homologous locus. Antimicrob Agents Chemother. outubro de 2008;52(10):. 3597-603.
- 39.Lamb D C, Kelly D E, Waterman M R, Stromstedt M, Rozman D et al.Characteristics of the heterologously expressed human lanosterol 14 alpha-demethylase (other names: P45014DM, CYP51, P45051) and inhibition of the purified human and Candida albicans CYP51 with azole antifungal agents. , Yeast 15(9), 755-63.
- 40.Korashy H M, Shayeganpour A, Brocks D R, AOS El-Kadi.Induction of cytochrome P450 1A1 by ketoconazole and itraconazole but not fluconazole in murine and human hepatoma cell lines. Toxicol Sci. maio de 2007;97(1):. 32-43.
- 41.Taxvig C, Vinggaard A M, Hass U, Axelstad M, Metzdorff S et al.Endocrine-disrupting properties in vivo of widely used azole fungicides. , Int J Androl. abril de 31(2), 170-7.
- 42.Pont A, Williams P L, Azhar S, Reitz R E, Bochra C et al.Ketoconazole blocks testosterone synthesis. Arch Intern Med. novembro de 1982;142(12):. 2137-40.
Cited by (1)
- 1.do Nascimento Francisca BA, Valente Sá Lívia GA, de Andrade Neto João B, Cabral Vitória PF, Rodrigues Daniel S, et al, 2023, Antifungal activity of cisatracurium against fluconazole-resistantCandidaisolates and its antibiofilm effects, Future Microbiology, 18(10), 649, 10.2217/fmb-2022-0224