
Abstract
Isolation methods that employ readily-available inexpensive supplies on the open market, which are reliable, as well as economical, such as nucleic acid amplification techniques (NAAT) based on microfluidic technology in low-resource research settings (LRRS) that meets the ASSURED guidelines are essential to develop a noninvasive diagnostic colon cancer screen in stool using micro(mi)RNA molecules. A combination of a microfluidic-based MiRNA stool test with a reliable rolling circle amplification/detection method applied to the quantification of miRNA molecules, result in an affordable sensitive and specific isothermal method for the noninvasive quantitative detection of miRNAs in LRRS.
Scientists and engineers have become interested in miRNAs, and they have intensified their efforts to apply emerging simple detection tools to the important bioanalytical challenge of quantifying these small 18-26 nt long molecules. Some of the proposed approaches incorporate novel material, such as simple centrifuges and methods based on microfluidic technology, while others utilize the interesting biological properties of these molecules, such as forming branched RCA structures, allowing for the detection of these biomarker molecules at an attomolar "aM" concentration level, using low cost extraction and isothermal amplification methods in LRRS.
We have been interested in studying colorectal cancer (CRC) because it is the 3rd most common malignancy worldwide, and stool can be obtained noninvasively from the patients. We have focused in this research on colon cancer (CC) because it is more common in the USA than rectal cancer (RC). The innovation of our approach lies in the exploratory use of an affordable, quantitative miRNA profiling in noninvasive stool samples in LRRS, whose extracted fragile total RNA is stabilized shortly after excretion from stool by commercially available kits, so it does not ever fragment, followed by quantitative standardized analytical tests that are neither labor intensive, nor require expensive instrumentation, in order to develop apanel of novel miRNA genes for the noninvasive diagnostic screening of early left and right sporadic colon cancers, more economically, and with higher sensitivity and specificity than any other colon cancer screening test currently available on the market.
To show the clinical sensitivity and specificity of the proposed quantitative miRNA test using simple methodologies in LRRS,the miRNA results are to be correlated with FOBT, colonoscopy, and pathology data. Standardization establishes test’s performance criteria (sample selection, optimal sample running conditions, preservation and storage), in order to ensure that the assay will perform the same way in any laboratory, by any trained personnel, anywhere in low-resource laboratory settings worldwide.
Author Contributions
Academic Editor: Murat Kara, Mugla Sitki Kocman University School of Medicine, Department of Medical Genetics 48000 Kotekli, Mugla,Turkey
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2019 Farid E Ahmed, et al.
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have no conflicts of interest to declare.
Citation:
Introduction
Nucleic acid amplification techniques (NAAT) are becoming an increasingly important part of clinicians’ tool box. Nucleic acid detection today has mainly been confined thus far to wealthy, developed countries or to large centralized facilities in the developing world that can afford resources required to carry out these methods.
Economic and infrastructural realities dictate that diagnostics for the low-resource settings (LRS) need to be affordable, sensitive, specific, user-friendly, rapid, equipment-free and deliverable to end users (ASSURED) 1. NAAT assays that use quantitative polymerase chain reaction (qPCR) amplification are capable of providing excellent sensitivity and specificity within about 60 min, but generally fail to meet the ASSURED guidelines for simplicity, affordability, rapidity and robustness, equipment-free operation and deliverability because of the need for precise temperature control (use of a thermocycler), skilled personnel and very clean conditions, making it challenging to utilize qPCR in many LRS 2. Recently, there have been significant developments in a class of NAATs that does not require temperature cycling 3. These isothermal amplification techniques use a variety of reaction principles to specifically amplify nucleic acids (NAs) through isothermal melting, exponential amplification and intermediate target generation, and in some cases targets can be detected directly without an instrument, as in loop-mediated amplification (LAMP) 4. Moreover, these NAATs have been shown to be less sensitive to inhibitors than PCR, and in some cases no power or instruments are required, and reagents may be stored in reaction tubes with sufficient stability, thus avoiding the need for a cold-chain during delivery and storage. Use of engineered phase change materials (EPCMs) --such as calcium oxide (CaO) with a temperature set point of 55oC±1 oC as a heat source for LAMP-- to manufacture exothermal chemical heating units has produced consistent and constant-temperature incubators for isothermal NA amplifications that are suitable for a variety of isothermal techniques 5.
A toe warmer (Heat Factory, Vista, CA) was used as a heat source for a helicase-dependent isothermal amplification (HAD), 65oC±2oC of DNA for 55 min. It consists of a polypropylene bag containing iron, salt, activated carbon, cellulose and vermiculite. When exposed to oxygen in the air, the iron powder oxidizes in an exothermic reaction. The salt serves as a catalyst; the carbon serves to disperse the heat; vermiculite is used as an insulator to maintain the generated heat; and cellulose is a filler. Since the chemical components inside the commercially available toe warmer are fixed, the duration of the reaction as well as the degree of heat generated will mainly depend on air exposure and humidity supply. Relative humidity is important because water on the surface of iron particles can enhance iron oxidation and therefore generate more heat. When this toe warmer is put inside a covered Styrofoam cup that has holes in it, the amount of vapor that interacts with iron particles is dependent on the number of holes on the Styrofoam cup; the more holes, the more vapor can be trapped to react with iron. Thus, by controlling the number of holes in the cup, the temp and duration of the reaction can be manipulated 6.
Microfluidic-enabled testing is an option in the development of appropriate, easy-to-adapt diagnostic technologies in LSR. It affords several advantages such as low cost, energy efficiency, capacity to perform complex functions in a single device, lightweight and portability for in-field testing, high sensitivity with small sample volumes and a relatively fast output 7. These characteristics make microfluidics a natural fit for portable point-of-care diagnostic systems. A common approach for making diagnostic technologies a feasible option in LRS is to make them completely self-contained and/or purely disposable. In addition to being simple and reliable, they must be robust enough so that little maintenance is needed, and operations can occur at a wide range of ambient temperatures (10 to 40oC), and the device should be functional within the infrastructure of a LSR 8. Hence, the development and use of isothermal amplification devices/ assays in LRS is an attractive option.
MicroRNAs are short endogenous noncoding RNAs (16-26 nucleotides) that have been associated with cancer 9. Several unique characteristics of miRNAs, including their small size, sequence homology among family members and low abundance in total RNA samples make them difficult to analyze. Northern blot is the standard method for analysis, but it has low sensitivity, is time consuming and requires large amount of samples 10. Locked nucleic acid (LNA) hybridization probes were applied to traditional Northern blotting protocols to assuage several of the aforementioned problems 11. Incorporation of digoxigenin into complementary RNA strands used to visualize Northern blots and an accompanying chemiluminescent readout have reduced the time-to-results from days to hours, and increased the shelf-lives of the probes compared to radioactively labeled strands 12.
Cloning was one of the first methods used to detect miRNAs 13, but it is not suited for hightroughput analysis. A more recent development has been mirage:miRNA serial analysis of gene expression 14.
Microarrays allowed simultaneous analysis of multiple samples, but lacked sensitivity & specificity 15. Recent amplification strategies on microarrays using fluorescence 16 or nanoparticles 17 required complex protocols.
Homogenous methods such as RT-qPCR 18, modified invader assay 19 and ribozyme-based signal amplification 20 have been attempted; however, the short length of miRNAs make their experimental design very sophisticated, and doubly fluorescence-labeled or LNA probes make detection expensive. Electrochemical detection of miRNAs showed a detection limit at 650 fm 23. Rolling circle amplification (RCA) using various probes, several amplification enzymes and a variety of amplification methods has been developed for the ultrasensitive detection of miRNAs, to 1fM 24, 25, 26, 27, and is employed in this research to refine and enhance its application & utility for an affordable/sensitive isothermal detection of miRNA in LRS, as detailed below.
The biomarker discovery approach outlined herein has been designed to test the hypothesis that “quantitative measurement of the expression of a carefully-selected panel of few miRNA genes in stool is a reliable, sensitive and specific diagnostic indicator for early non-invasive screening of colon cancer (CC)”. To prove this hypothesis, it must first be validated in a study, as proposed herein. While scientists may differ about the merits of using a prospective versus a retrospective design, we will use a nested case control epidemiology design that employs a prospective specimen collection, retrospective blind evaluation (PRoBE) of healthy controls and test colon cancer patients 54, as specifically delineated by the National Cancer Institute’s (NCI’s) Early Detection Research Network (EDRN) http://edrn.nci.nih.govfor cancer biomarker discovery studies.
The innovation of the presented approach lies in “the exploratory use of an affordable, quantitative miRNA profiling in easily obtained noninvasive stool samples, whose extracted fragile total RNA is stabilized shortly after stool excretion by commercially available kits so it does not ever fragment, followed by quantitative analytical tests that are neither labor intensive, nor require expensive instrumentation, to initially develop a panel of novel miRNA genes for the diagnostic screening of early left and right sporadic colon cancer more economically, and with higher sensitivity and specificity than any other colon cancer screening test currently available in LRS”.
Materials and Methods Needed for this Approach
Standardize Sample Acquisition, Processing and Handling; Justify Number of Selected Subjects & Epidemiologically Randomize Selection of Study Population
Acquisition of Patients and Specimens for Analysis
Collaborating clinicians must be made aware of the constraints imposed by working with RNA, and the need to preserve it so it does not ever fragment after extraction of these short ~20 nucleotide-long molecules from human stool. After consenting prospective individuals when they report to the clinic for consult, those individuals (age 18 to 90 years old) not showing polyps, or inflammatory bowel disease such as colitis or diverticulitis, will be asked by their physicians if they wish to participate in the study. If they agree, they or their guardian will be consented, each given a stool collection kit and detailed oral and written collection instructions. Each study subject will collect one 10 g stool sample, in a standardized fashion, in a large 40 cc plastic jacket prior to any bowel preparation. The study nurse will show and ask participants to obtain samples using a clean plastic spoon from both the mucinous layer, which is rich in colonocytes, and the non-mucinous parts of stool in order to have a representation of the entire colon (both right and left side colon) 33, 51, 55, to be preserved in sterile urine vials overnight at room temperature in the fixative RNALater® (Invitrogen) added at 2.5 ml per 1 g of stool, followed by calling the laboratory personnel to pick up the sample by next morning. Preserved samples will be stored in our labs at -80oC in small aliquots until needed. When ready for analysis, samples will be defrosted at room temp, filtered through a nylon mesh by laboratory personnel at GEM Tox facilities in order to remove the preservative and any debris prior to extraction of total RNA. All laboratory work is carried out and standardized under blind conditions and, in accordance with our institute’s Standard Operating Procedures (SOPs) for the handling of biohazardous material.
MiRNA data are to be analyzed by RCA, and using 80% power in a “Power Analysis” calculation to detect differences in expression between CC stages and control group at a larger standard deviation and a 75% reduction in the difference, to optain adequate group size of control subjects and colon cancer patients from different stages of colon cancer (adenomas, TNM stage 0-I; TNM stage II; and TNM stages III & IV) selected randomly is an adequate number of samples to test. for the purpose of this study.
Randomized Selection of Control Human Subjects and Case Patients
An adequate number of subjects who do not have colon cancer need to be chosed randomly.To avoid bias, and ensure that biomarker selection and outcome assessment does not influence each other, a prospective specimen collection retrospective blinded evaluation (PRoBE) design randomized selection 54 of control subjects and case patients from consented cohort population are chosen without knowing a priori who has what diagnosis, and will want to collect the stool specimen prior to removal of the lesion, in addition to collected consenting specimens on patients undergoing colonoscopy to form a suitable cohort to randomly selected the appropriate number of colon cancers. Then adenoma cases are matched 1 to 1 to the cancer cases for age (+/- 5 years), gender, clinic and month of diagnosis. Similarly, the normal controls from among the collected specimens are matched to the cancer and adenoma cases. If there is no match, the data are liberalized to allow +/- 2 months, in order to have collected a case-case-control group nested in the overall colonoscopy cohort that is collected. The quantitative miRNA analysis by RCA is then carried out on all coded samples at once, with the investigators blind to knowledge of the patients’ diagnosis, so that no analytical bias is introduced. While there may be some volunteer bias present, which may affect the studied miRNA markers, it is recommended to collecte demographic and clinical data on both groups (those who participated and those who did not) and compare for the following factors: age, gender, race/ethnicity, reason for colonoscopy, diagnoses, so that an assessment can be made at the conclusion of the study as to what degree selection may have affected study results.
A six aims and a proposed timeline for achieving these aims in a 5 year clinical research study are detailed in Table 1 below:
Table 1. Proposed timeline for accomplishing research aims during the 5 years study period| Method-Aim/Months | Aim 1:Standardize sample acquisition,processing and handling & epidem- iologically selectstudy population | Aim 2: Standardize total RNA extraction by QA methods & perform RCAs to study miRNAs gene expression at various CC stages | Aim 3: Employ epigenetics to study genetic heterogeneity by identifying MSS-MSI phenotypes & investigate promoter methylation in CC stages | Aim 4: Finalize accessing test performance characteristics & numerical under-pinning of the proposed RCA approach for CC | Aim 5: Use statistical methods for data analyses | Aim 6: Provide & carry out alternate standardized methods to achieve study goals, if needed |
| 1-4 | ·········a | · | ||||
| 5-8 | ········ | · | · | |||
| 9-12 | ······ | ·· | · | ·· | · | |
| 13-16 | ········· | · | ||||
| 17-20 | ·· ···· ·· | · | · | |||
| 21-24 | ······ | ·· | · | ·· | · | |
| 25-28 | ········· | · | ||||
| 29-32 | ········ | · | · | |||
| 33-36 | ······ | ·· | · | ·· | · | |
| 37-40 | ·········a | · | ||||
| 41-44 | ········ | · | · | |||
| 45-48 | ······ | ·· | · | ·· | · | |
| 49-52 | ········· | · | ||||
| 53-57 | ·· ···· ·· | · | · | |||
| 57-60 | ······ | ·· | · | ·· | · |
a· = Refers to potential frequency and/or level of effort needed to accomplish/complete project aim.
1. Standardize sample acquisition, processing and handling; justify number of selected subjects & epidemiologically randomize selection of study population.
2. Standardize total RNA extraction from stool colonocytes by strict quality assurance (QA) criteria, and perform Rolling Circle Amplification (RCA) to study microRNAs gene expression at various stages (0- IV) of colon cancer (CC) progression.
3. Employ epigenetic methods to study genetic heterogeneity by identifying MSS & MSI phenotypes, as well as investigating promoter methylation in miRNA genes at various stages of CC Progression
4. Finalize accessing test performance characteristics of the proposed RCA diagnostic approach for colon cancer.
5. Use statistical methods for data analyses.
6. Provide and carry out alternate standardized technical methods for achieving the above aims in the unlikely event that the proposed approach, or the outlined methods fail to achieve study goals.
Extraction of Standardized Total RNA from Stool Colonocytes by Strict Quality Colonocytes by Strict Quality Assurance (QA) Criteria
RNA isolation procedures (both automatic and manual), compared to those used for isolation of DNA from stool samples 54, 72, 73, 74, 75, 76, 77, 78, are standardized and made simple by us using improved commercially available reagents and kits 33, 51, 79 to extract high quality total RNA from an environment as hostile as stool 80; thus, shattering the myth that it is difficult to employ RNA as a screening substrate. The trick is to stabilize total RNA shortly after obtaining fresh stool by fixing samples in a chaotropic agent and observing that RNA does not ever fragment thereafter. Fragmented RNA results in poor cDNA synthesis, and ultimately in less than optimal RCA amplification.
Total RNA can be manually isolate from colon laser capture microdissected (LCM) tissue, and from stool by using Qiagen’s RNeasy Isolation Kit® (Qiagen, Valencia, CA) containing a RLT buffer (a guanidinium-based solution) according to manufacturer’s instructions, getting the advantage of manufacturer’s established validation and quality control standards, thereby increasing the probability of good results. Generally, total RNA isolated from stool is suitable for amplification of miRNAs by RCA without the need to further purify mRNA because purified mRNA involves additional steps, and the increased sensitivity could be balanced for by the potential loss of material 67.
Isolated RNA in nuclease-free water can be stored at -80oC until needed. It can then be quantified spectrophotometrically at 260 nm. Acquiring sufficient mRNA to analyze from stool or isolated colonocytes is feasible, as each cell contain ~ 20 pg total RNA, and only few nanograms are needed per RCA reaction 81.
Preparation of dsDNA template
A total of 50 pmol of forward and reverse oligonucleotides (Invitrogen), according to the sequence of the interrogated miRNA, can be annealed by incubation at 75oC for 5 min, and then slowly cooled to room temperature (~ 30 min). The fill-in reaction to form dsDNA template can be performed in a 20 µl volume containing 10 mM Tris-HCl, 50 mM NaCl, 10 mM MgCl2, 1 mM DTT, 0.25 mM dNTPs and 5U Klenow Fragment (3’ – 5’ exo) New England Biolabs at 37oC for 1 h. Then the reaction mixture can heated at 75oC for 20 min to inactivate the enzyme, and slowly cooled to room temperature for dscDNA annealing (see Figure 1 A, upper level).
In Vitro Transcription Reaction
A total of 20 µl of the above dsDNA template mixture can be added into 30 µl of in vitro transcription buffer containing 0.5 mM NTPs, 40 mM Tris-HCl, 6 mM MgCl2, 10 mM dithiothreitol (DTT), 2 mM spermidine, 100 U ribonuclease inhibitor and 50 U T7 RNA polymerase (New England Biolabs). The reaction can be run at 37oC for 4 h, then 1 U of RNase-free DNase I is added to digest DNA template, followed by purification of precursor miRNAs in transcription mixture with phenol/chloroform extraction. Transcript precursor miRNAs can be examined by running 2% agarose gel, and the concentration is determined spectrophotometrically at a wave length of 260 nm.
Phosphorylation and Reverse Transcriptase (RT) Reaction
Before miRNA is reverse transcribed, the RT primer is to be chemically phosphorylated at the 5’ end by heating a 0.5 nmol RT primer to 75oC for 5 min, then chilling on ice prior to treatment with kinase. The 50 µl reaction volume containing 50 mM Tris-HCl (pH 8.0), 10 mM MgCl2, 5 mM DTT, 2 mM DTT, 1 mM ATP, 20 U T4 polynucleotide kinase and DNA probe formation can be carried out at 37oC for 2 h, followed by inactivation of the T4 polynucleotide kinase at 65oC for 20 min.
RT reaction for formation of cDNA from miRNA can be carried out in a 10 µl reaction volume that contains 1µl of total RNA sample, 500 nM 5’-phosphorylated RT primers, 20 U MultiScribeÔ RT (Applied Biosystems, Foster City, CA) , 50 µM dNTPs and 1X reaction buffer (pH 8.3, 50 mM Tris-HCl, 75 mM KCl and 3 mM MgCl2), followed by the addition of 2.5 U Ribonuclease H at 37oC for 20 min for the degradation of miRNA strand in the RNA-DNA hybrid. These steps can be followed up by probe ligation and rolling circle amplification (see Figure 1 B and Figure 2).
Figure 1.Principle of isothermal ramification amplification (RAM) for sensitive and real-time quantitative analysis of miRNA. (A) This assay that is based on threshold cycle (CT) principle. It has three coordinated steps: 1. Reverse transcription of miRNA, 2. C-Probe ligation, and 3. Ramification amplification. (B) Dynamic range and sensitivity of Synthetic let-7a (from 103 to 1010 copies per reaction, 10 nM to 1 fM) 26.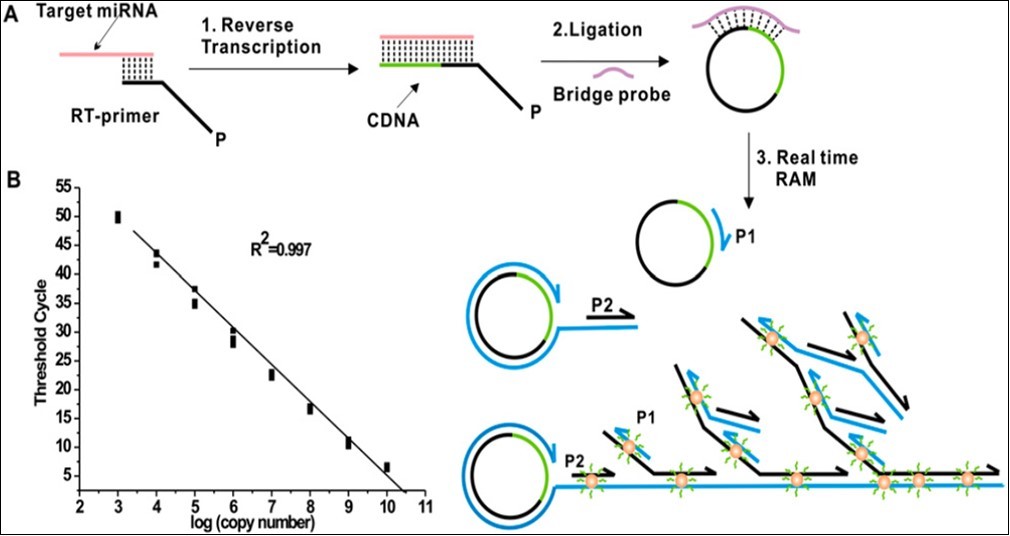
Figure 2.Principle of a miRNA detection system based on padlock probe recognition of miRNAs and rolling circle amplification (RCA). (A) Padlock probes are linear DNA probes where terminal sequences are designed to specifically recognize and hybridize to two adjacent sequences of a particular miRNA. (B) The padlock probes annealing to the perfectly matching miRNA termplate are circularized upon addition of DNA ligase. (C) After ligation the annealed miRNA serves as a primer for linear rolling circle amplification by a phi29 DNA polymerase. (D) The phi29 DNA polymerase facilitates rolling circle amplification, thereby producing a DNA product containing multiple copies of the miRNA sequence from 24.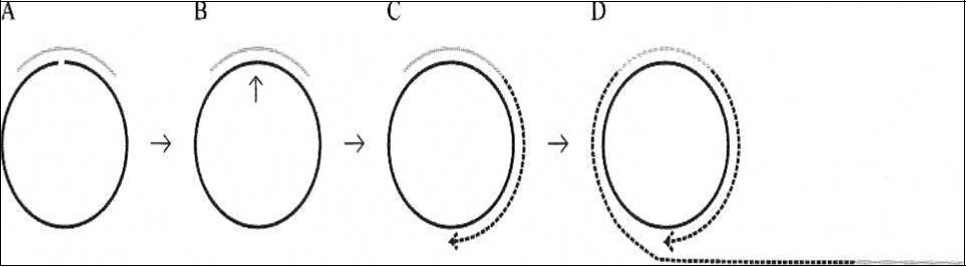
Ligation Followed by Rolling Circle Assay
Jonstrup et al24 (Figure 2) developed a linearly amplified simple detection system by using miRNA as a template to cyclize padlock probes as a primer for RCA, achieving a detection limit of ~ 10 pm. However, while the method is simple, linear amplifications by itself cannot satisfy the requirements for high detection sensitivity. A T7 exonuclease-assisted cyclic enzymatic (CEAM) amplification is based on nucleases in which one target leads to many cycles of target dependent nuclease cleavage of reporter probes for output signal amplification. Exonuclease III (Exo II), for example, catalyzes the stepwise removal of mononucleotides from the 3’-blunt or recessed terminus of duplex DNA. This unique property of Exo III-based CEAM allowed it to be widely used for optical (fluorescence, UV-Visible, SPR) and also for electrical amplifications for the detecting DNA, proteins and small molecules 82. However, because of the linear nature of the amplification process, it was able to only achieve detection limits in the pico molar (pM) range. Additionally, the limited intrinsic properties of the nucleases used have made earlier CEAMs not applicable to miRNA targets 83.
Genomic DNA Extraction and MSI Analysis
For extraction of genomic DNA from stool in a LRS, a manual extraction for stool samples that employs a QIAamp DNA Stool Mini Kit (Qiagen, Valencia, CA), can be used.
A fast, convenient method for label-free detection of amplified markers, which can be routinely carried out, using genomic DNA isolated from stool colonocytes by the DNA Extraction Kit (Qiagen), on 6 of the 10 Bethesda markers recommended by NCI 122. These include: three mononucleotide repeats (BAT-25, -26 and -40) and three dinucleotide repeats (D2S123, D5S346 and D17S250), which have shown a sensitivity of >96%, and a specificity of >99% for identifying tumors with MMR deficiency. The presence of MSI-H correlates with an older age of diagnosis, the presence of tumors in the proximal colon and female sex. A cut of 30% for markers has been recommended to use for classifying tumors with MSI-H 104. Label-free amplification with reagents found in the Gold Taq Kit (Applied Biosystems) can be used, as details in references 123, 124. Upon completion of the non-PCR amplifications in foam cups or boxes, the products of each individual sample can be processed by on-chip electrophoresis using a DNA 1000 Lab Chip in the Agilent 2100 Bioanalyzer 125, 126.
DNA methylation analysis by MethylLight technology
Modifying the genomic DNA before non-PCR amplification by sodium metabisulfite treatment 127 can be employed, followed by DNA recovery using a modified Qiagen Viral RNA Mini Kit 128, and eluted DNA samples can be stored at -80oC until needed. Methyl Light analysis can performed by fluorescence-based assay, using β-actin as a reference normalization gene. The % of fully methylated molecule at a specific locus, called PMR, is then calculated 129, 130. SAS software can be used to determine the statistical significance of the results. The two major molecular CC subgroups (MSS and MSI) can be studied in selected miRNAs, at various stages of CC progression in stool samples as a complex molecular signature using factor analysis (FA) 131, combined with linear discriminate analysis (LDA) 132 to identify the cancer’s molecular characteristics.
Use Statistical Methods for Data Analyses, and Driving a Predictive MiRNA Index (PMI)
If the difference in miRNA gene expression between healthy and cancer patients and among the stages of cancer at the end of the study is as large and as informative for multiple miRNA genes, suggesting that classification procedures could be based on values exceeding a threshold, then sophisticated classification would not be needed to distinguish between the two study groups. However, if inconsistent differences on large samples are found, then it is recommended to use predictive classification methods, as detailed below.
The goal in predictive classification will be to assign cases to predefined classes based on information collected from the cases. In the simplest setting, the classes (i.e., tumors) are labeled cancerous and non-cancerous. Statistical analyses for predictive classification of the information collected (i.e., microarrays and qPCR on miRNA genes) attempt to approximate an optimal classifier. Classification can be linear, nonlinear, or nonparametric 140, 141. The miRNA expression data are to be analyzed first with parametric statistics such as Student t-test or analysis of variance (ANOVA) if data distribution is random, or with nonparametric Kruskall-Wallis, Mann-Whitney and Fisher exact tests, if distribution is not random 142. If needed, complicated models as multivariate analysis and logistic discrimination 143 can also be employed.
False positive discovery rates (expected portion of incorrect assignment among the expected assignments) can also be assessed by statistical methods 144, 145, 146, 147, as it could reflect on the effectiveness of test because of the need to do follow up tests on false positives. The number of optimal miRNA genes needed to achieve an optimum gene panel for predicting carcinogenesis in stool can also be established by statistical methods.
For the corrected index, cross-validation 148 can be used to: protect against overfitting, address the difficulties with using the data to both fit, assess the fit of the model, and determine the number of samples needed for a cancer study, where the expected proportion of genes’ expression common to two independently randomly selected samples is estimated to be between 20% and 50% 144. Efron and Tibshirani 149 suggested dividing the data into 10 equal parts and using one part to assess the model produced by the other nine; this is repeated for each of the 10 parts. Cross-validation provides a more realistic estimate of the misclassification rate. The area under the ROC curves, in which sensitivity is plotted as a function of (1 – specificity), can be employed to describe the trade-off between sensitivity and specificity 150, 151.
Power analysis can be implemented for estimating sample size in such a study 152. Moreover, power analysis, as well as first and second order validation studies can be carried out to access the degree of separation and reproducibility of our data 153.
Principal component analysis (PCA) 154, which is a multivariate dimension reduction technique to simplify grouping of genes that show aberrant expression from those not showing expression, or a much reduced expression, can be employed. In cases where several genes by themselves appear to offer distinct & clear separation between control or cancer cases in stool samples, a predictive miRNA Index (PMI) may not be needed.
If by the end of the study, the miRNA gene panel (or a derived PMI) is better than existing screening methods, all of the data generated can be used to assess the model so over-fitting is not a concern. The level of gene expression can be displayed in a database using parallel coordinate plots 155, 156 produced by the lattice package in R (version 2.9.0, The R Foundation for Statistical Computing, http://cran.r-project.org), and S-plus software (Insightful Corporation, Seattle, WA). Other packages such as GESS (Gene Expression Statistical System) published by NCSS (www.ncss.com) can also be employed, with each subject having his or her medical record number as the key ID for merging various tables in the database.
If results show that individual miRNA genes offer distinct and clear separation between control and cancer, there will be not need to derive a predictive miRNA index (PMI) 157, 158. It may, however, be necessary to do so if data were not supportive. In this case, results of the quantitative expression of the miRNA genes can be used to determine a cutoff for a positive result. PMI results above or below the cutoff are to considered positive or negative, respectively, in all subsequent studies. Resulting data can then be then used to check the sensitivity and specificity of the index using a two by two matrix (Table 2) 159. If the sensitivity falls below 90% or the specificity falls below 95%, forthcoming data using additional miRNA genes can then be used with linear or logistic discriminant analysis to refine the index.
% Sensitivity = TP x 100
TP + FN
% Specificity = TN x 100
FP + TN
Table 2. Predictive MicroRNA Index (PMI)| Cancer Cases | Tue Positive (TP) | False Negative (FN) |
| Normal Subjects | False Positive (FP) | True Negative (TN) |
Alternate Standardized Technical Methods for Achieving the Above Research Aims
Above methods represent the most practical, least labor-intensive and economical approach to accomplish research aims. However, in few problematic samples (<5%) in control, or pre-malignant or malignant cases, it may be necessary to use other methods. However, because the error rate is so small and would occur in control and cases, adopting different extraction/analyses methods will not bias results.
Manual Extraction of Total RNA from Problematic Samples Using the AGPC Method
In very few samples, inhibitors present in stool may make it difficult to isolate RNA using Qiagen kits that provide the advantage of manufacturer's established validation and QC standards. In such cases, RNA can be manually isolated by a modification of the classical acid guanidinium thiocyanate-phenol-chloroform (AGPC) method 160, using chaotropic guanidinium thiocyanate (GSC) that inactivates ribonucleases and most microorganisms.
Use of a Plate Assay to Study MicroRNA Expression
Signosis, Inc., Sunnyvale, CA (www.signosisinc.com) introduced high throughput plate assay for monitoring individual miRNAs, without the need to carry out a RT reaction. In that assay one of the bridge oligos partially hybridizes with the miRNA molecule and the capture oligo, and another bridge forms a hybrid between the miRNA molecule and the detection oligo. The hybrid that is sensitive to the miRNA sequence is immobilized onto a plate and detected by a streptavidin-horse radish peroxidase conjugate and chemiluminescent substrate using a plate reader. One oligonucleotide difference prevents hybrid formation; thus miRNA isoform could be differentiated.
Carry out miRNA Measurements from Exosomes and Microvessicles Extracted from Stool
MiRNAs are resistant to ribonucleases present in stool, probably by inclusion in lipid or lipoprotein complexes 161 in either microvessicles (up to 1 µm), or in small membrane vesicles of endocytic origin known as exosomes (50-100 nm) 162. The mechanism of release of miRNAs from exosomes and microvesicles is unclear, although an apoptotic delivery candidate is shed from cells during apoptosis 163. Exosomes released from human and murine mast cell lines were shown to contain mRNAs and miRNAs 164. MiRNAs in microvessicles regulate cellular differentiation of blood cells and certain metabolic pathways, and modulate immune functions 165. MiRNA signatures of tumor-derived exosomes function as diagnostic markers in ovarian cancer, and tumor-derived miRNA profiles are not significantly different from exosomal miRNA profiles 166. Exosomal miRNAs was extracted from stool colonocytes by differential centrifugation, followed by filtration through 0.22 µm filters to remove cell debris, total RNA extracted by Trizol & concentration measured spectrophotometrically at λ 280 167.
Conclusions
Cui et al27 combined RCA with CEAM developing a dual amplification method for the ultrasensitive detection of miRNAs with excellent sensitivity, and successfully applied it to differentiate the let-7a expression levels of hepatoma cells and normal hepatocytes, demonstrating the potential application of this method for early cancer diagnosis. Ligations can be performed by taking 10 µl of RT product and 1µl of the probe to be employed in such research, such as linear molecular beacons (LMBs), which have been specifically designed for the CEAM method (Figure 3). The figure shows that RCA-CEAM contains circular template, phi29 DNA polymerase, dNTPs, and CEAM reaction substrates (exonuclease, report probe). To improve hybridization kinetics and reduce background signal, specially-designed linear molecular beacons (LMBs) have been used as the report probe 84. A LMB is a linear oligonucleotide probe with a fluorophore and a quencher attached to the terminal and penultimate nucleotides, respectively. The circular template of RCA was designed to embed the complementary sequences of target miRNA and reporter LMB 85.
Figure 3.Principle of rolling circle amplification combined with a T7 exonuclease-assisted cyclic enzymatic (RCA-CEAM) dual amplification method for highly sensitive detection of miRNA molecules 27.
In the first step, binding of target miRNA to the circular template permits replication of the circular template with the help of phi DNA polymerase. RCA, being an isothermal nucleic acid (NA) amplification process produces a long ssDNA with numerous copies of the complementary sequence of the original circular template, consisting of many repeat units that are complementary to the LMB, and can hybridize to thousands of these LMB molecules
In the second step, using Exo III-- that has extra residual exonuclease activity on ssDNA-- results in reduced sensitivity due to a high background signal. On the other hand, replacing Exo III with a T7 exonuclease (T7 Exo), which hydrolyzes mononucleotides from blunt or recessed 5’-termini of duplex DNA, but cannot work on ssDNA, results in the digestion of the LMB, and the separation of the fluorophore (FAM) from the quencher leading to fluorescence emission, followed by binding of each released unit to another LMB and initiation of a new cleavage process. Fluorescence was determined spectrofluorometrically. The excitation and emission wavelengths were set at 490 and 520 nm, respectively.
Through such a cyclic hybridization-hydrolysis process, each RCA product will have many units and each unit can induce the cleavage of a large number of LMBs to liberate numerous fluorophores, resulting in a highly efficient fluorescent signal amplification process. Moreover, since the RCA employs a short sequence miRNA as the primer, byproducts as the non-active, long-chain pri-miRNA, stem-loop structure pre-miRNA precursor, or mismatch miRNA will not trigger the RCA reaction. Thus, by combining two linear amplification processes, the resulting dual amplification method provided an excellent sensitivity (<12 fm) and selectivity for miRNA analysis, and therefore is used in this research for the sensitive & selective detection of miRNAs.
The performance of RCA using miRNA as the primer has been tested. As shown in Figure 4 A, with SYBR Green dye present in the reaction system, the fluorescence spectra showed a significant increase after addition of 10 nm miRNA. In a dual amplification step that tests the combined performance of RCA and CEAM, with increasing target concentration from 25 fM to 5 pM, a gradual increase in fluorescence intensity was also observed (Figure 4B). In Figure 4C, the fluorescence intensity change showed a linear positive correlation towards target miRNA concentration in the range between 25 fM to 1 pM, with a detection limit of less than 12 M. It is interesting that while the RCA by itself can only achieve a detection limit of about 10 pM, the RCA-CEAM is three orders of magnitude lower than that of conventional RCA, or other signal amplification methods. Therefore, the dual amplification method can easily distinguish the let-7 miRNA family (e.g., let-7e and let-7f, which differ from target let 7a by only one base), as shown in Figure 4D 27.
Figure 4.Sensitive and selective miRNA detection using the dual amplification method. (A) Fluorescence spectra before and after RCA reaction. (B) Fluorescence spectra of dual amplification method over a range of target miRNA concentrations. (C) The relationship between the fluorescence intensity change and target miRNA concentration. (D) The dual signal amplification assay differentials the let-7 family. From reference 27.
Use of Inexpensive Centrifuges and Disposable miRNA Amplification Platforms Suitable for LRS
Diagnostic technologies have been developed and have been applied in LRS. For example, Wong et al modified a hand-powered egg beater into a centrifuge to isolate human plasma from whole blood 86 (Figure 5), which can be employed in this research as a simple centrifuge suitable for us in LRS.
Figure 5.Plasma isolation using an egg beater centrifuge [86).
Lateral-flow tests are now widely used in testing of genetically modified organisms in food 87. However, due to several limitations, lateral flow assays cannot provide sufficient sensitivity and specificity required for accurate diagnosis in many cases 88. Several paper-based microfluidic devices have been reported for use in bioanalysis to detect glucose and protein, pH and alkaline phosphatase 89. LaBarre and his colleagues validated a first complete, non-instrumented nucleic acid amplification test (NAAT) for loop-mediated isothermal amplification assay (LAMP) made from a reusable $4 insulated soup thermos container 5.
Nucleic acid-based assays have been successfully implemented in recent years in many fields exploiting their rapid and accurate analysis in several areas such as medical diagnostics, forensics, environmental analysis and biodefense 90, 91. Standard, tube-based real-time qPCR can produce results within 30 min 92. However, the need for precise temperature control requiring use of a thermocycler or an alternate precise heat controller capable of functioning at a wide range of temperatures, skilled personnel and very clean conditions have made it challenging to utilize qPCR in many resource limited settings. Hence, the development of an affordable lower temperature, isothermal amplification device/assay has been an attractive option.
Affordable Microfluidic Chips for NA Amplification
A low throughput (LT) microfluidic chip (Figure 6 A) to be used in this kind of low-resource research can be constructed from six layers of cyclic olefin polymer (COP) film (Zeon Chemicals, Louisville, KY). The microfluidic channels are made by cutting 188 µm thick COP films using a Graft ROBO Pro CE5000-40 cutter plotter (Graphtec America Inc, Irvine, CA) 93. The two sheets with inlet and outlet holes and the four sheets of cut films are stacked and thermally bonded in a hot press (Heated Press 4386, Carver, Wabash, IN) at 125oC for 20 min with 1.5 tons of pressure followed by 10 min of curing at 137oC without any pressure. The inlet and outlet bridge channels are 188 µm high, 200 µm wide and 10 mm long. The reaction chamber measuring 0.75 x 2.5 x 15 mm can hold a helicase-dependent isothermal amplification (HAD) reaction with a volume of 25 µl 6.
Figure 6.Microfluific devises employed in this research. (A) Low throughput (LT) device. (B) High throughput (HT) device showing three sections: sample preparation column, RT chamber and above them are PCR channels. Plug to seal inlet and outlet of the device is shown in the background.
A high throughput (HT) microfluidic chip (Figure 6 B) that can be used in such research can be fabricated in thermoplastic Zeonex 690R (Zeon Chemicals, Louisville, KY) by hot-embossing a blank 0.7 mm thick Zeonex plaque using an epoxy master mold and then thermally sealing the imprinted channel with a 0.7 mm blank Zeonex cover slip 94. Three open ports in the chip were fitted with nanoports (Upchurch Scientific, Oak Harbor, WA) using epoxy glue to facilitate loading the specimens, adding reagents and collecting amplification products. The entire chip is 70 mm in length, 25 mm in width and 1.4 mm in height.
The integrated chip consists of three functional chambers: a solid phase extraction (SPE) column for sample preparation and RNA extraction 95, an RT chamber, and amplification channels as shown in Figure 6 B. The SPE column was made with a mixture of 16% v/v ethylene dimethacrylate, 24% butyl metacrylate, 42% 1-dodecanol, 18% cyclohexanol and 1% photoinitiator 2-dimethylamino-4-(methyl-phenylamino)-phenol. Silica microspheres were added into the monolith solution in a 1:1 ratio (v/v).
The monolith was cross linked by UV irradiation using an Ultraviolet Crosslinker (CL-1000, UPV Inc, Upland CA) for 5 min through both sides of the chip. Silica microspheres (0.7 µm, Polysciences, Inc) were prepared by centrifugation at 6,600 rpm for 5 min, aspirating the suspension solution and drying on heat block overnight to remove excess water. The inner channel surface was grafted with 97% v/v methylacrylate and 3% v/v benzophenone, UV irradiated for 10 min at λ 265 nm in an Ultraviolet Crosslinker as described above to facilitate covalent attachment between the SPE column and the inner plastic channel surface.
Reagents for SPE column fabrication can be purchased from Sigma (St Louis, MO). Before fabrication of the SPE column, the channel was cleaned with 50 µl RNAse Away (Molecular BioProducts, San Diego, CA) and then rinsed with 100 µl nuclease free water. Dr. Catherine M Klapperich of Department of Boston University Mechanical Engineering Department, Boston, MA provided us with prototypes of these devices to try during the data generating phase of the study.
In the proposed research, we have simplified making these microfluidic devices by methods such as using sharp blades for cutting the thermoplastic polymers, using double adhesive surfaces for binding polymers, instead of employing a hot press to make these microfluidic devices more amenable to manufacturing in LRS.
Styrofoam Cup or Box as a Container for Housing the Microfluidic Devices
As seen in Figure 7 and Figure 8, the microfluidic devices are housed in a Styrofoam cup or box, for the low-throughput and high throughput devices, respectively. If reactions are carried out at room temperature, nothing else will be needed. For reactions requiring 37oC, liquid sodium acetate will be used as heat source. In a purified form, at typical ambient temperatures, liquid NaAc is thermodynamically unstable, but kinetically stable due the absence of nucleating sites for crystal formation. The application of a mechanical shock, like dropping thermometer in the container initiates exothermic crystallization, and when mixed with a 25% aqueous solution, the phase change occurs repeatedly at ~ 37oC 5.
Figure 7.Styrofoam cup containing the LT microfluidic device and a thermometer
Figure 8.Styrofoam box containing the HT microfluidic device and a thermometer
In this system, NaAc acts as both the exothermic reactant and the engineered phase change material (EPCM) 96. This system has the advantage of being regenerable (for example, to immersion of the NaAc in boiling water) and is the preferred system for isothermal amplification methods operating at temperatures below 45oC, as well as for other diagnostic applications requiring heating below 45oC 97. For temperatures of ~ 55oC, CaO can be used 5.
For amplifications requiring a temperature of 65oC, a toe warmer containing iron, salt, activated carbon, cellulose and vermiculite can be used as a heating source due to the oxidization of the iron powder in an exothermic reaction 6.
Temperature control during a given experiment can be governed by a number of 1 mm diameter vent holes punched on the opposite sides of the Styrofoam cup or box. The more holes, the more vapor particle can be trapped to react with iron. Therefore, by controlling the number of holes on the Styrofoam cup or box, the temperature and duration of the reaction can be manipulated.
A thermocouple attached to the microfluidic reaction chamber, instead of a thermometer, can also be used to monitor the microfluidic reaction temperature.
Why Use a MicroRNA Assay for a Diagnostic Colon Cancer Screening
Review of Worldwide scientific literature has shown that for colon cancer screening, miRNA markers are more comprehensive and preferable to a DNA- 55, epigenetic-, mRNA- 51, a protein-based marker 56, or to a FOBT test 57. An added advantage of the use of the stable, nondegradable miRNAs by RCA-based methods is the potential for using a simple amplification platform consisting of polymer-based microfluidic chips and disposable Styrofoam cups, which makes this platform much more economical and more easily acceptable by laboratory personnel performing these assays in low-resource settings.
Suitability of Stool for Developing Highly Sensitive Diagnostic Biomarker to Screen for Colon Cancer
Links between miRNAs and CRC have been reported in several studies in cells in culture, blood or tissue 58, 59; colon cancer cell lines 60, 61; colon adenocarcinoma tumors 62; stage II colon cancer 63; serum of CRC patients 64; blood of colon cancer patients 65; and human stool 33, 66, 67.
Stool testing has several advantages over other colon cancer screening methods as it is truly noninvasive and requires no unpleasant cathartic preparation, formal health care visits, or time away from work or routine activities. Unlike sigmoidoscopy, it reflects the full length of the colorectum and samples can be taken in a way that represents the right and left side of the colon. It is also believed that colonocytes are released continuously and abundantly into the fecal stream, contrary to blood that is released intermittently as in guaiac FOBT 68, 69; therefore, this natural enrichment phenomenon partially obviates the need to use a laboratory technique to enrich for tumorigenic colonocytes, as for example when blood is used for testing. Furthermore, because testing can be performed on mail-in-specimens, geographic access to stool screening is essentially unimpeded. The ACS has recognized stool-based molecular testing as a promising screening technology for CRC (www.cancer.org).
Our results and others have shown that the presence of bacterial DNA, non-transformed RNA and other interfering substances in stool does not interfere with measuring either mRNA or miRNA expression 33, 66, 70. Besides, stool colonocytes contain much more miRNA and mRNA than that available in free circulation such as in plasma 65, 66, 70. To further ensure that we are choosing human and not bacterial genes, we have sequenced ten samples at the start of the research using whole-transcriptome RNAseq analysis from small amounts of extracted total RNA 71.
Genetic Instability and the Role of Epigenetics in Colon Cancer Heterogeneity
Three major forms of genetic instability in colon cancer have been described based on the presence or absence of functional DNA mismatch repair (MMR) 98, 99, 100, contributing further to the enormous heterogeneity of CC.
a) In about 15-20% of sporadic CRC cases and in most patients with HNPCC/Lynch syndrome, tumors with defective mismatch (dMMR) leads to the presence of a phenotype termed microsatellite instability (MSI) 101, characterized by the absence of protein expression of any one of several genes involved in DNA repair, including hMLH1. hMSH2, hMSH6 or hPMS102. MSI is recognized by the presence of insertion and deletion mutations in replicative DNA sequences termed microsatellites, which consist of repeating mononucleotide, dinucleotide, or polynucleotide sequence loci. In sporadic CRC, three distinct MSI phenotypes have been characterized: i) MSS (none of the loci demonstrate instability and is believed to have evolved through the classical adenoma-carcinoma sequence, ii) MSI with low instability (MSI-L; from < 30 to 40% of loci), and iii) MSI with high instability (MSI-H; from 30% to > 40% of loci) 103, mostly due to inactivation of hMLH1 gene 104.
b) In about 40% of colon cancers that are characterized by epigenetic changes, especially cytosine DNA hypermethylation, a phenomenon termed “CpG Island Methylator Phenotype”, CIMP 105, 106, is associated with the 5’ regulatory end and spanning the promoter region of almost all housekeeping genes, as well as half of tissue-specific genes, leads to genetic instability and mutation 107, 108. It is a reversible process catalyzed by three major DNA methyltransferases (DNMT1, DNMT3a and DNMT3b) 109. By combining mutation with epigenetic analyses, 3 subclasses of CC with distinct clinicopathological features were identified (CIMP1, CIMP2 and CIMP negative) 110. Other critical epigenetic marks are chemical modification of N-terminal tails of chromatin protein histone through complex postranslational modifications such as lysine acetylation, arginine and lysine methylation or serine phosphorylation. MSI/CIMP+ve and MSS/CIMP-ve are a subset of proximal CCs believed to have evolved through flat lesions that include both adenomas and serrated polyps 111.
c) In the remaining 45% of CCs, chromosomal instability (CIN) such as aneuploid or polyploid karyotypes leads to translocations (gains and losses of large segments of chromosomes) 112; but will not be studies in this project.
Colon tumors show differential expression of miRNAs depending on mismatch repair status. MiRNA expression in colon tumors has an epigenetic component, and altered expression that may reflect a reversion to regulatory programs characteristic of undifferentiated proliferative developmental states 113.
MiRNAs also undergo epigenetic inactivation 110, and miRNA expression in CRC is associated with MSI subgroups 114, 115. MiRNAs may affect chromatin structure by regulating key histone modification; for example, cartilage-specific miR-140 targets histone deacetylase 4 in mice 116, and miRNAs may be involved in meiotic silencing of unsynapsed chromatin in mice 117. In addition, DNA methylation enzymes DNMT1, 3a and 3b were predicted to be potential miRNA targets 118. Moreover, a specific group of miRNAs (epi-miRNAs), miR-107, -124a, -127, directly target effectors of the epigenetic machinery such as DNMTs, histone deacetylases and polycomb repressive complex genes, and indirectly affect the expression of suppressor genes 119, 120, 121. The challenge has been to identify those driver methylation changes that are critical for tumor initiation, and distinguish these from methylation changes that are merely passenger events that accompany the transformation process but that have no effect per se on carcinogenesis. In this study we will look at genetic instability in stool of CC patients.
Promoter Methylation
We studies promoter methylation using MethylLight analysis for 11 miRNAs: 7 genes showing increased expression (miR-1991-3p, miR-196a, miR-96, miR-214, miR-21, miR-201, miR-106a), and 4 showing decreased expression (miR-1275p, miR-222, miR-146a miRNA-143) in genomic DNA extracted from stool of 20 individuals (4 normal, 4 TNM stage 0-I, 4 stage II, 4 stage III and 5 stages IV) by the sensitive MethylLight method 164. The various genes varied to some extent with respect to their sensitivities to promoter methylation, with some values ≥ 60% for stage IV (miRNA-199a-3p, miRNA-96, miRNA-214, miRNA-21), some in the 30s% in stage IV CC (miRNA-106a, miRNA-146a, miRNA-143), and some in between (miRNA-196a, miRNA-20a, miRNA-127-5p, miRNA-222) (Table 3).
Table 3. DNA promoter methylation analysis of miRNA genes in stool by Methyl Light technology| Gene/Stage | % | Control | Stage 0-I | Stage II | Stage III | Stage IV |
| miR-199a-3p | % Sens. | 5 | 29 | 49 | 55 | 65 |
| % Spec. | 93 | 89 | 87 | 90 | 92 | |
| miR-196a | % Sens. | 4 | 35 | 46 | 50 | 58 |
| % Spec. | 91 | 95 | 93 | 96 | 94 | |
| miR-96 | % Sens. | 6 | 39 | 47 | 53 | 69 |
| % Spec. | 89 | 86 | 89 | 87 | 90 | |
| miR-214 | % Sens. | 3 | 33 | 44 | 52 | 60 |
| % Spec. | 93 | 79 | 88 | 90 | 92 | |
| miR-21 | % Sens. | 6 | 52 | 56 | 58 | 61 |
| % Spec. | 90 | 88 | 91 | 93 | 89 | |
| miR-20a | % Sens. | 3 | 41 | 45 | 50 | 54 |
| % Spec. | 94 | 89 | 93 | 96 | 91 | |
| miR-106a | % Sens. | 5 | 16 | 24 | 30 | 35 |
| % Spec. | 89 | 90 | 92 | 94 | 91 | |
| miR-127-5p | % Sens. | 7 | 27 | 42 | 46 | 51 |
| % Spec. | 93 | 90 | 91 | 94 | 94 | |
| miR-222 | % Sens. | 5 | 19 | 30 | 39 | 46 |
| % Spec. | 92 | 90 | 93 | 91 | 92 | |
| miR-146a | % Sens. | 8 | 13 | 29 | 33 | 38 |
| % Spec. | 88 | 84 | 86 | 89 | 90 | |
| miR-143 | % Sens. | 4 | 11 | 22 | 29 | 33 |
| % Spec. | 86 | 87 | 89 | 88 | 90 |
Accessing Test Performance Characteristics (TPC) of the MicroRNA Approach, and Providing Numerical Underpinning of the RCA Method as a Function of Total RNA
Isothermal amplification values of the miRNA gene panel in stool and colonocyte samples of normal subjects and CC patients can be compared to IHC FOBT test and colonoscopy results obtained from patients’ medical records in the 140 study subjects to access TPC of the miRNA approach.
False positive discovery rates (expected proportion of incorrect assignment among the accepted assignments) can be assed statistically 133, 134, as it could reflect on the cost effectiveness of our test because of the need to do follow up tests on false positives. The number of optimal miRNA genes needed for correct diagnosis of colon cancer was established by statistical method detailed below.
Cytological methods Can be used on purified colonocytes isolated from all stages of CC, employing Papanicolaou and Giemsa staining as described for CRC 135 to provide numerical underpinning of the RCA method as a function of total RNA. These methods have shown sensitivity for detecting tumor cells in smears comparable to that found in biopsy specimens (78.1% versus 83.66%) 136. The numerical method can be accomplished by isolating a known number of colonocytes from 1g stool (from both normal and neoplastic preparations), extracting total RNA from them to determine the actual concentration of total RNA per 1 g of stool sample, running RCA and determining the RCA value of selected miRNA genes in our system, arriving at an average quantitative RCA value per a certain quantity (pg or ng) of total RNA. Colonocytes were isolated from stool using magnetic separation as we detailed before 137, 138. Then comparing the miRNA expression profile to that of total RNA extracted from whole stool could be construed as a validation that the pattern observed in stool is truly due to the presence of tumor cells therein. Some exsosomal RNA 139 could be released from purified colonocytes into stool, and we have corrected for that effect.
In situ hybridization analysis in tissue microarray (TMA) for miR-21 (Figure 9) can be performed after digestion in protease; the tissue and probe miRNA (1 pmol/µl, 5’ digoxigenin-tagged; Exiqon A/S, Vedbaek, Denmark) were co-incubated at 60oC for 5 min, then hybridized for 15 h at 37oC.
Figure 9.Differential expression of miR-21 in colon cancer versus the adjacent normal tissue. In situ hybridization analysis for miR-21 in this core tissue microarray sample showed very high expression in the cytoplasm of cancer dells (right part of the image) and very low expression in the adjacent normal colonic tissue epithelia (left part of image). The signal is blue due to nitroblue tetrazolium and bromochlorindol phosphate and counterstaining with nuclear fast red, x10 67.
After a wash in 0.1X of standard saline citrate (SSC) solution and 2% bovine serum albumin (Sigma-Aldrich, St Louis, MO) at 50oC for 10 min, the miRNA-probe complex can be visualized via nitroblue tetrazolium and bromochlorindol phosphate (NBT/BCIP) (Roche Molecular Biochemicals, Indianapolis, IN) appears blue due to the action of the alkaline phosphatase that is conjugated to the antidigoxigen in antibody. The figure show high differential expression of the miRNA in the cytoplasm of cancer dells (right part of the image) and low expression in the adjacent normal colonic tissue epithelia (left part of image). The signal is seen in blue due to NBT/BCIP staining, and counterstaining with nuclear fast red 67.
Cytological methods on purified colonocytes that employed Giemsa stain showed a sensitivity for detecting tumor cells in stool smears of 80% for higher stages of CC (stages 3 and 4), which is slightly better than what was reported earlier (i.e., about 78%) 65.
Numerical underpinning of the miRNAs as a function of total RNA can be carried out on colonocytes isolated from stool 165 before any preservative was added to five healthy control samples, and five TNM stage IV colon cancer samples, extracting total RNA from them and determining the actual amount of total RNA per stool sample, and from the average CP values, taking into account that some exsosomal RNA will not be released from purified colonocytes into stool, and arbitrarily corrected for that effect 66. It is evident from our preliminary data shown in Table 4 that an average CP value for stage IV colon carcinoma of 21.90 is invariably different from a CP value of 26.05 for healthy controls.
Table 4. Numerical Underpinning of miRNA markers as a function of total RNA.| Patient # | Diagnosis | Total RNA(µg/g of stool) | CP* | |
| Sample | Group | |||
| 2 | Healthy control | 0.28 | 25.99 | |
| 5 | Healthy control | 0.3 | 27.21 | |
| 8 | Healthy control | 0.29 | 25.96 | 26.05 |
| 14 | Healthy control | 0.31 | 25.09 | |
| 17 | Healthy control | 0.32 | 26.01 | |
| 56 | Stage IV carcinoma | 0.31 | 20.18 | |
| 57 | Stage IV carcinoma | 0.32 | 22.03 | |
| 58 | Stage IV carcinoma | 0.31 | 22.43 | 21.9 |
| 59 | Stage IV carcinoma | 0.3 | 21.37 | |
| 60 | Stage IV carcinoma | 0.31 | 23.49 | |
Test performance characteristics (TPC) of the miRNA approach obtained by the CP values of the miRNA genes calculated from stool colonocyte samples of normal healthy individuals and patients with colon cancer were compared to the commonly used FOBT test and with colonoscopy results obtained from patients’ medical records in 60 subjects (20 control subjects and 40 colon cancer patients with various TNM stages). The data showed high correlation with colonoscopy results obtained from patients’ medical records for the controls and colon cancer patients studied.
Significance of the Employed Technology for MiRNA Detection in Colon Cancer
It is important to develop reliable and economical nucleic acid amplification techniques (NAAT) & other isolation/separation methods, using readily-available inexpensive instruments and affordable microfluidic-based technologies in low-resource settings (LRS) that meets the ASSURED (Affordable, Sensitive, Specific, User-friendly, Rapid and robust, Equipment-free, and Deliverable to end user) guidelines published by WHO for diagnostic devices for developing countries 1. We have been working on developing reliable diagnostic methods for screening methods for colon cancer during the past 20 years using mRNA and micro(mi)RNA molecules.
We are interested in colorectal cancer (CRC) because it is the 3rd most common malignancy worldwide, with an estimated one million new cases and half million deaths yearly 28. Epidemiologic, anatomical, cellular and molecular evidence suggest that colon cancers (CCs) and rectal cancers (RCs) represent two distinct types of tumors 29. We have focused our research on employing colon cancer because it is more abundant in the USA, and much more CC patients report to our clinics compared to RC 30, 31, 32, which will provide researchers all needed CC samples to achieve study aims in reasonable time. We are in the opinion that a combination of basic centrifuges and microfluidic-based miRNA stool test with a reliable rolling circle amplification/detection method results in developing of an affordable sensitive and specific isothermal method for the non-expensive and noninvasive diagnostic screening of colon cancer in LRS.
Innovation of the Proposed MicroRNA Approach
The discovery of small noncoding protein sequences, 17-27 nucleotides long RNAs microRNAs (miRNAs), has opened new opportunities for a non-invasive test for early diagnosis of many cancers 33. There are miRNA sequences from 271 organisms; 38,589 hairpin precursors, and 48,860 mature miRNAs in the October 2018 http://mirbase.org34. MiRNA functions seem to regulate development 35 and apoptosis 36, and specific miRNAs are critical in oncogenesis 37, effective in classifying solid 38, 39, 40, 41, 42 & liquid tumors 43, 44, and serve as oncogenes or suppressor genes 45. MiRNA genes are frequently located at fragile sites, as well as minimal regions of loss of heterozygosity, or amplification of common breakpoint regions, suggesting their involvement in carcinogenesis 46. MiRNAs have great promise serving as biomarkers for cancer diagnosis, prognosis and/or response to therapy 33, 47, 48, 49. Profiles of miRNA expression differ between normal tissues and tumor types, and evidence suggests that miRNA expression profiles can cluster similar tumor types together more accurately than expression profiles of protein-coding mRNA genes 9, 50.
Unlike screening for large numbers of messenger (m)RNAs, a modest number of miRNAs is used to differentiate cancer from normal 51, and unlike mRNA, miRNAs in stool remain largely intact and stable for detection 33. Therefore, miRNAs are better molecules to use for developing a reliable noninvasive diagnostic screen for colon cancer, since we have found out during our earlier studies that: a) the presence of Escherichia coli does not hinder detection of miRNA by a sensitive technique such as RCA, and b) the miRNA expression patterns are the same in primary tumor, or diseased tissue, as in stool samples 79.
In the past twenty years, physical scientists and engineers, as well as biomedical scientists have become increasingly interested in miRNAs, and have intensified efforts to apply emerging separation/detection tools to these important and bioanalytically challenging small molecules. Some of these approaches incorporate novel material and reagents such as use of manual rotation devices as centrifuges, and use of microfluidic technology, while others have utilized the interesting biological properties of miRNAs --such as forming branched RCA structure-- allowing for the quantitative detection of these biomarker molecules at an "aM" concentration level using low cost extraction and isothermal amplification methods in LRS 6, 24, 52, 53.
Quantitative Milestones to be Accomplished
There has been three principal milestones to be achieved by the end of this research study:
Milestone 1: Derivation of Workable miRNA Gene Panel, or Alternatively a PMI in Stool Based on RCA
This milestone is considered to have been achieved, if 81 (90%) of the 90 patients with CC cancer has a miRNA panel that gave a numerical RCA value in stool by a microfluidic method.
Milestone 2: Accessing TPC & Providing Numerical Underpinning of the Method as a Function of Total RNA
Test performance characteristics (TPC) of the miRNA approach can be determined by comparing quantitative RCA values of miRNA genes obtained from stool samples of normal subjects and colon cancer patients with IHC FOBT test and with colonoscopy results obtained from patients’ medical records in the 90 colon cancer patients.
A numerical underpinning of the method can determined by calculating the amount of total RNA in 1 g of stool and determining the average RCA value for the miRNA gene(s) per a known amount (pg or ng) of total RNA.
Milestone 3: Establishing the Clinical Sensitivity and Specificity of miRNA Gene Panel, or a PMI
a) Perform IHC FOBT in parallel with the miRNA panel for each stool sample of the 140 enrolled study subjects (50 healthy controls and 90 CC patients).
b) Review colonoscopy results, and check blinded histopathologic reports of biopsies/surgical specimens and final patients’ diagnosis, including those carried out on removed polyp biopsies, from patients’ medical records.
c) From the RCA results of the genes’ panel selected (or a PMI) obtained from stool samples of normal and cancer patients, construct a 2 x 2 table (Table 2) was to determine the clinical sensitivity and specificity of the miRNA assy.
d) The calculated sensitivity/specificity of the miRNA assay are then compared to the FOBT method and colonoscopy results obtained from patients’ medical records to establish TPCs. If the results are at least as specific as the FOBT (95%), and the sensitivity is ≥90%, which exceeds colonoscopy, then this milestone is considered to have been achieved.
References
- 1.Mabey D, R W Peeling, Ustianowski A, M D Perkins. (2004) Diagnostics for the developing world.Nat Rev Microbiol2:. 231-240.
- 2.Morshed M G, Lee M K, Jorgensen D, Isaac-Renton J L. (2007) Molecular methods used in clinical laboratory: prospects and pitfalls. , Fems Immun Mol Microbiol 49, 184-192.
- 3.Andersen D, M von Nickisch-Rosenegk, Bier F F. (2009) Helicase-dependent amplifications: use in a chip amplification and potential for point-of cure diagnostics. Exp Rev Mol Diagnostics9:. 645-650.
- 4.Tomita N, Mori Y, Kanda H, Notomi T. (2008) Loop-mediated isothermal amplification (LAMP) of gene sequences and simple visul detection of products.Nat. Protocols3: 877-882.
- 5.LaBarre P, K R Hawkins, Gerlach J, Wilmoth J, Beddoe A et al. (2011) A simple, inexpensive device for nucleic acid amplification without electricity–toward instrument-free molecular diagnostics in low-resource settings.PLoS ONE:. 19738.
- 6.Huang S, Do J, Mahalanabis M, Fan A, Zhao L et al. (2013) Low cost extraction and isothermal amplification of DNA for infectious diarrhea diagnosis.PLoS ONE8: e60059.
- 8.Yager P, Edwards T, Fu E, Nelson K, M R Tam et al. (2006) Microfluidic diagnostic technologies for global public health.Nature442:. 412-418.
- 9.Lu J, Getz G, Miska E A, Alvarez-Saavedra E, Lamb J et al. (2005) MicroRNA expression profiles classify human cancers.Nature435:. 834-838.
- 10.Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. (2001) Identification of novel genes coding for small expressed RNAs.Science294:. 853-858.
- 11.Varallyay E, Burgyn J, Navelda Z. (2008) MicroRNA detection by northern blotting using locked nucleic acid probes.Nat. Protoc3: 190-196.
- 12.S H Ramkissoon, L A Mainwaring, E M Soland, N S Young, Kajigaya S. (2006) Nonisotopic detection of microRNA using gigoxigenin labelled RNAprobes.Mol Cell. Probe20: 1-4.
- 13.Lagos-Quintana M, Rauhut R, Yalin A, Meyer J, Lendeckel W et al. (2002) Identification of tissue specific microRNA from mouse. , Cancer Biol 12, 735-739.
- 14.J M Cummins, He Y, R J Leary, Pargliarini R, Jr Diaz L A et al. (2006) Szufranska A E, Labourier E. Proc Natl Acad Sci USA 103: , Raymond C K, Rhuts B S, Juhl H, Kinzler K W, Voglstein B and Velculesca V E 3687-3692.
- 15.Lee I, Ajay S S, Chen H, Maragama A, Wang N et al. (2008) Discriminating single-base difference miRNA expression using microarray probe.Nucl Acids Res36:. 27.
- 16.Wang H, R A Ach, Curry B. (2007) Direct and sensitive miRNA profiling from low input total RNA.RNA13:. 151-159.
- 17.Gao Z, Yang Z. (2006) Detection of microRNA using electrocatalytic nanoparticle tags.Anal. Chem78: 1470-1477.
- 18.Chen C, Ridzon D A, Broomer A J, Zhou Z, Lee D H et al. (2005) Real-time quantification of microRNAs by stem-loop RT-PCR. Livak KJ and Guegler KJ , Nucleic Acids Res 33, 179.
- 21.J S Hartig, Grune I, S H Najafi-, Famulouk M. (2004) Sequence-specific detection of microRNA by signal-amplyfying ribozymes.Am Chem Soc126:. 722-723.
- 22.Zhang Y, Li Z, Cheng Y. (2008) Amplified fluorescence determination of microRNA in homogenous solution with cationic conjugated polymers. , Chem Commun 48, 6579-6581.
- 23.Sun Y, K J Gregory, N G Chen, Golovlev V. (2012) Rapid and direct microRNA quantification by an enzymatic luminescence assay.Anal. Biochem429: 11-17.
- 24.H V Tran, Piro B, Reisberg S, Anquetin G, H T Due et al. (2014) An innovative strategy for direct electrochemical detection of microRNA biomarkers.Anal Bioanl Chem406:. 1241-1244.
- 25.S P Jonstrup, Koch J, Kjens J. (2006) A microRNA detection system basede on padlock probes and rolling circle amplification.RNA12:. 1747-1757.
- 26.Cheng Y, Zhang X, Li Z, Jiao X, Wang Y et al. (2009) Highly sensitive determination of microRNA using target-primed branched rolling circle amplification.Angew Chem Int Ed48:. 3268-3272.
- 27.Yao B, Li J, Huang H, Sun C, Wang Z et al. (2009) Quantitative analysis of zeptomole microRNAs basede on isothermal ramification amplification.RNA15:. 1787-1794.
- 28.Cui L, Zhu Z, Lin N, Zhang H, Guan Z et al. (2014) A T7 exonuclease-assisted cyclic enzymatic amplification method coupled with rolling circle amplification a dual amplification strategy for sensitive and selective microRNA detection.Chem. Commun50: 1576-1578.
- 29.Ahmed F E. (2006) Gene-gene, gene-environment & multiple interactions in colorectal cancer.J. , Env Sci HealthC 24, 1-101.
- 30.Cheng L, Eng G, Nieman L Z, Kapadia A S, Du X L. (2011) Trends in colorectal cancer incidence by anatomic site and disease stage. in the United States from 1976 to 2005.Am J Clin Oncol34: 573-580.
- 31.Wu X, Chen V W, Martin J. (2004) Subsite-specific colorectal cancer incidence rates and stage distributions among Asians and. Pacific Islanders in the United States,1955 to , Cancer Epidemiol Biomarkers Prev 13, 1215-1222.
- 32.Frattini M, Balestra D, Suardi S, Oggionni M, Alberici P et al. (2004) Different genetic features associated with colon and rectal carcinogenesis.Clin Cancer Res10:. 4015-4021.
- 33.Birkenkamp-Demtroder K, Olesen S H, SÆrensen F B, Laurberg S, Laiho P et al. (2003) Differential gene expression in colon cancer of the ceacum versus the sigmoid and rectosigmoid.Gut54:. 374-384.
- 34.Ahmed F E, Jeffries C D, Vos P W, Flake G, Nuovo G J et al. (2009) Diagnostic microRNA markers for screening sporadic human colon cancer and ulcerative colitis in stool and tissue.Cancer Genom. Proteom6: 281-296.
- 35.Kozomara A, Birgaoanu M, Griffiths-Jones S. (2019) miRBase: from microRNA sequences to function.Nucleic Acids Res47:. 155-162.
- 36.Reinhart B J, Slack F J, Basson M, Pasquinell A E, Bettinger J C. (2000) RNA regulates developmental timing in Caenorhabditis elegans. Rougvie AE, Horvitz HR and Ruvkun G: , Nature 403, 901-906.
- 39.Cummins J M, Velculescu V E. (2006) Implication of microRNA profiling for cancer diagnosis.Oncogene15:. 6220-6227.
- 40.Lee E J, Gusev Y, Jiang J, Nuovo G J, Lerner M et al. (2007) Expression profiling identifies distinct microRNA signature in pancreatic cancer.Int. , J Cancer120: 1046-1054.
- 41.Yanaihara N, Caplen N, Bowman E, Seike M, Kumamoto K et al. (2006) Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Groce CM and Harris CC , Cancer Cell 9, 189-198.
- 42.Iorio M V, Ferracin M, Liu C G, Veronese A, Spizzo R et al. (2005) MicroRNA gene expression deregulation in human breast cancer.Cancer. Res65: 7065-7070.
- 43.Cummins J M, He Y, Leary R J, Pagliarini R, Diaz LA Jr et al. (2006) . The colorectal microRNome.Proc Natl Acad Sci USA103: 3687-3692.
- 44.Calin G A, Ferracin M, Cimmino A, Dileva G, Shimiz M et al. (2005) A microRNA signature associated with prognosis and progression in chronic lymphocytic leukemia.N. , Eng J Med353: 1793-1801.
- 45.Eis P S, Tam W, Sun L, Chadburn A, Li Z et al. (2003) . Accumulation of miR-155 and BIC RNA in human B cell lymphomas.Proc Natl Acad Sci USA102: 3627-3632.
- 47.Calin G A, Sevignai C, Dumitru C D, Hyslop T, Noch E et al. (2004) Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers.Proc Natl Acad Sci USA101:. 2999-3004.
- 48.Schepler T, Reinert J T, Oslenfeld M S, Christensen L L, Silahtaroglu A N et al. (2008) Diagnostic and prognostic microRNAs in Stage II colon cancer.Cancer. Res68: 6416-6424.
- 49.Barbarotto E, Schmittgen T D, Calin G A. (2008) . MicroRNAs and cancer: Profile, profile, profile.Int J Cancer122: 969-977.
- 50.Schetter A J, Leung S Y, Sohn J J, Zanetti K A, Bowman E D et al. (2008) MicroRNA expression profile associated with progression and therapeutic outcome in colon adenocarcinoma.J Am Med Assoc299:. 425-436.
- 52.Ahmed F E, Vos P, iJames S, Lysle D T, Allison R R et al. (2007) Transcriptomic molecular markers for screening human colon cancer. in stool & tissue.Cancer Genom Proteom4: 1-20.
- 53.Qavi A J, Kindt J J, Bailey R C. (2010) sizing up the future of microRNA analysis.Anal Bioanal Chem398:. 2535-2549.
- 54.Stougaard M, Juul S, Andersen F F, Knudsen B R. (2011) Strategies for highly sensitive biomarker detection by Rolling Circle Amplification of signals from nucleic acid composed sensors.Integr. Biol3: 982-9920.
- 55.M S Pepe, Feng Z, Janes H, Bossuyt P M, Potter J D. (2008) Pivotal evaluation of the occurance of a biomarker used for classification or prediction: standards for study design. , of Cancer.J Natl Cancer Inst100: 1432-1438.
- 56.Itzkowitz S H, Jandorf L, Brand R, Rabeneck L, S et al. (2007) Improved fecal DNA test for colorectal cancer screening.Clin Gastroenterol Hepatol5:. , Markowitz S and Shuber A 111-117.
- 57.Ahmed F E. (2009) Liquid chromatography-mass spectrometry: A tool for proteome analysis & biomarker discovery and validation.Exp Opin Mol Diag3:. 429-444.
- 58.Wada R, Miksushima T, Shiratori Y. (2006) Comparison of the immunochemical fecal occult blood test and total colonoscopy in the asymptomatic population.Gastroenterology125:. 422-428.
- 59.Barbarotto E, Schmittgen T D, Calin G A. (2008) . MicroRNAs and cancer: Profile, profile, profile.Int J Cancer122: 969-977.
- 60.Shi B, Stepp-Lorenzino L, Prisco M, Linsley P, deAngelis T et al. (2007) . MicroRNA 145 targets the insulin receptor substrate-1 and inhibits the growth of colon cancer cells.J Biol Chem282: 32582-32590.
- 61.Michael M Z, O’Connor S M, NG van Holst Pellekaan, Young G P, James R J. (2003) Reduced accumulation of specific microRNAs in colorectal neoplasia.Mol Cancer Res1:. 882-891.
- 62.Lu M, Zhang Q, Deng M, Miao J, Guo Y et al. (2008) An analysis of human microRNA and disease associations.PLoS ONE3: e3420.
- 63.Schetter A J, Leung S Y, Sohn J J, Zanetti K A, Bowman E D et al. (2008) MicroRNA expression profile associated with progression and therapeutic outcome in colon adenocarcinoma.J Am Med Assoc299:. 425-436.
- 64.Schepler T, Reinert J T, Oslenfeld M S, Christensen L L, Silahtaroglu A N et al. (2008) Diagnostic and prognostic microRNAs in Stage II colon cancer.Cancer. Res68: 6416-6424.
- 65.Chen X, Ba Y, Ma L, Cai X, Yin Y et al. (2008) Characterization of microRNAs in serum: a novel class of biomarkers for diagnosis of cancer and other diseases.Cell. Res18: 997-1006.
- 66.Ahmed F E, Ahmed N C, Vos P, Bonnerup C, Atkins J N et al. (2012) Diagnostic microRNA markers to screen for sporadic human colon cancer in blood.Cancer Genom. Proteom9: 179-192.
- 67.Link A, Balaguer F, Shen Y, Nagasaka T, Lozano J J et al. (2010) Fecal microRNAs as novel biomarkers for colon cancer screening.Cancer Epidemiol Biomarkers Prev19:. 1766-1774.
- 68.Ahmed F E, Ahmed N C, Vos P W, Bonnerup C, Atkins J N et al. (2013) Diagnostic microRNA markers to screen for sporadic human colon cancer in stool: I. Proof of principle.Cancer Genom. Proteom10: 93-114.
- 69.Kronborg O, Fenger C, Olsen J, Jorgensen O D, Sondergaard O. (1996) Randomized study of screening for colorectal cancer with faecal-occult-blood test.Lancet348:. 1467-1471.
- 70.Jorgensen O D, Kronborg O, Fenger C. (2002) A randomized study of screening for colorectal cancer using faecal occult blood testing: results after 13 years and seven biennial screening rounds.Gut50:. 29-32.
- 71.Davidson L A, Lupton J R, Miskovsky E, Fields A P, Chapkin R S. (2003) Quantification of human intestinal gene expression profiling using exfoliated colonocytes: a pilot study.Biomarkers8:. 51-61.
- 72.Tariq M A, Kim H J, Jejelowo O, Pourmand M. (2011) Whole-transcriptome RNAseq analysis from minute amounts of total RNA.Nucleic Acids Res39:. 120.
- 73.Traverso G, Shuber A, Levin B, Johnson C, Olsson L et al. (2002) . Detection of APC mutations in fecal and DNA from patients with colorectal tumors.N Engl J Med346: 311-320.
- 74.Imperiale T F, Ransohoff D F, Itzkowitz S H, Turnbull B A, Ross M A. (2004) Fecal DNA versus fecal occult blood for colorectal cancer screening in an average-risk population.New Eng J Med351:. 2704-2714.
- 75.Ahlquist D A, Skoletsky J E, Boynton K A, Harrington J J, Mahoney D W et al. (2000) Colorectal cancer screening by detection of altered human DNA in stool: feasibility of a multitarget assay panel. Thibodeau SN and Shuber AP , Gastroenterology 119, 1219-1227.
- 76.Lees N P, Hrrison K L, Hall C N, Morgison G P, Povey A C. (2007) Human colorectal mucosal O6-alkylguanine DNA-alkyltransferase activity and DNA-N7-methylguanine levels in colorectal adenoma cases and matched referents.Gut56:. 318-320.
- 77.Huang L H, Li L H, Yang F, Wang J F. (2007) Detection of aberrant methylation in fecal DNA s a molecular screening tool for colorectal cancer and precancerous lesions.World. , J Gastroenterol13: 950-954.
- 78.Model F, Osborn N, Ahlquist D, Gruetzmann R, Molnar B et al. (2007) Identification and validation of colorectal neoplasia—specific methylation markers for accurate classification of disease.Mol Cancer Res5:. 153-163.
- 79.Sidaransky D, Tokino T, Hamilton S R, Kinzly K W, Levin B et al. (1992) Identification of ras oncogene mutations in the stool of patients with curable colorectal tumors.Science256:. 102-105.
- 80.F E Ahmed, iJames S, Lysle D L, Dobbs LJ Jr, Johnke R M et al. (2004) Improved methods for extracting RNA from exfoliated human colonocytes in stool and RT-PCR analysis.Dig Dis Sci49:. 1889-1898.
- 81.Willis M S, Fuda F S, Chenault C B. (2002) Current methods of colorectal cancer screening.Med Lab Obser34:. 12-19.
- 82.Kuecker S J, Jin L, Kulig E, Oudrago G L, Roche P C et al. (1999) Analysis of PRL, PRL-R, TGFb1, and TGFb-RII gene expression in normal neoplastic breast tissue after laser capture microdissection.Appl Immunol Molec Morph7:. 193-200.
- 83.Cui L, Ke G, Lin X, Song Y, Zhang H et al. (2013) Cyclic enzymatic amplification method (CEAM) based on exonuclease III for highly sensitive bioanalysis.Methods63:. 202.
- 84.Cui L, Lin X, Lin N, Song Y, Zhu Z et al. (2012) Graphene oxide-protected DNA probes for multiplex microRNA analysisin complex biological samples based on cyclic enzymatic amplification method.Chem. Commun48: 194-196.
- 85.Yang C J, Cui L, Huang J, Yan L, Lin X et al. (2011) Linear molecular beacons for highly sensitive bioanalysis based on cyclic Exo III enzymatic amplification.BiosensBioelectron27:. 119-124.
- 86.Li J J, Chu Y, Lee B Y, Xie S X. (2008) Enzymatic signal amplification of molecular beacons for sensitive DNA detection. , Nucleic Acids Res 36, 36.
- 87.Wong A P, Gupta M, Shevkoplyas S S, Whiteside G M. (2002) Egg beater as centrifuge: isolating human blood plasma from whole blood in resource-poor settings.Lab on a. Chip8: 2032-2037.
- 89.Yager P, Edward Y, Fu E, Helton K, Nelson K et al. (2006) Microfluidic duiagnostic technologies for global public health.Nature442:. 412-418.
- 90.Martinez A W, Phillips S T, Whiteside G M, Garrillo E. (2009) Diagnostics for the developing world: microfluidic paper-based analytical devices.Anal. ChemB2: 3-10.
- 91.Sidransky D. (1997) Nucleic acid-based methods for the detection of cancer.Science278:. 1054-1058.
- 92.Kricka I J. (1999) Nucleic acid detection technologies: labels, strategies and formats.Clin. Chem45: 453-548.
- 93.GHW Onland, AJC van der Brule. (2005) Evaluation of conventional and real time PCR assays using two targets for confirmation of results of the COBAS AMPLICORChlamydia trachomatis/Nisseria gonorrhoeaetest for detection ofNisseria gonorrhoeaein clinical samples. , J Clin Microbiol 43, 2231-2235.
- 94.Bartholomeusz D A, Boutte R W, Andrade J D. (2005) Xurography: rapid prototyping of microstructures using a cutting plotter.Microelectrochemical Systems. J14: 1364-1374.
- 95.Bhattacharyya A, Klapperich C M. (2007) Mechanical and chemical analysis of plasma and ultraviolet ozone surface treatments for thermal bonding of polymeric microfluidic devices.Lab on a. Chip7: 867-882.
- 96.Bhattacharyya A, Klapperich C M. (2006) Thermoplastic microfluidic device for on-chip purification of nucleic acids for disposable diagnostics.Anal Chem78:. 788-792.
- 97.Weigl B, Domingo G, LaBarre P, Gerlach J. (2008) Towards non- and minimally instrumental, microfluidic-based diagnostic device.Lab on a. Chip8: 1994-2014.
- 98.Lai J J, Hoffan J M, Ebara M, Hoffman A S, Estournes C et al. (2007) Dual magnetic-/temperature-responsive nanoparticle for microfluidic separations and assays.Langmuir23:. 7385-7391.
- 99.Kalari S, Pfeifer G P. (2010) Identification of driver and passenger DNA methylation in cancer by epigenomic analysis.Adv. Genet70: 277-308.
- 100.Toyota M, Ohe-Toyota M, Ahuja N, JPJ Issa. (2000) Distinct genetic profiles in colorectal tumors with or without the CpG island methylator phenotype.Proc. , Natl Acad SciUSA 97, 710-715.
- 101.Weisenberger D J, Siegmund K D, Campan M, Young J, Long T I et al. (2006) CG island methylation phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer.Nat. Genet38: 787-793.
- 103.Herman J G, Baylin S B. (2003) Gene silencing in association with promoter hypermethylation.N. , Eng J Med349: 2042-2054.
- 105.Shen L, Toyota M.Kondo Yet al(2007) Integrated genetic and epigenetic analysis identifies three different classes of colon cancer.Proc Natl Acad Sci USA104:. 18654-18659.
- 106.Goldberg A D.Allis CD and Bernstein E (2007Epigenetics: a landscape takes shape.Cell128:. 635-638.
- 107.Jass J R. (2007) Molecular heterogeneity of colon cancer: implication for cancer control.Surg Oncol16:. 57-59.
- 108.Lengauer C, Kinzler K W, Vogelstein B. (1998) . Genetic instabilities in human cancer.Nature396: 643-649.
- 109.Feinberg A P, Vogelstein B. (1983) Hypomethylation distinguishes genes of some human cancers from their normal counterparts.Nature301:. 89-92.
- 110.Weber M, Hellmann I, Stadler M B. (2007) Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome.Nat. Genet39: 457-466.
- 111.NJM Raynal, Si J.Taby RF et al (2012) DNA methylation does not stably lock gene expression but instead serves as molecular mark for silencing memory.Cancer Res72:. 1170-1181.
- 113.Irizarry R A, Ladd-Acosta C, Wen.Bet al(2009) The human colon cancer methylome shows similar hypo- and hypermethylation at conserveds tissue-specific CpG island shores.Nat. Genet41: 178-186.
- 114.Hansen K D, Timp W.Corrada H et al (2011) Increased methylation variation in epigenetic domains across cancer types.Nature Genet43:. 768-775.
- 115.Sarver A L, French A J.Borralho PMet al(2009) Human colon cancer profiles show differential microRNA expression depending on mismatch repair status and are characteristic of undifferentiated proliferative states.BMC. , Cancer 9, 401.
- 116.JSL Earle, Luthra R.Romans Aet al(2010) Association of microRNA expression with microsatellite instability status in colorectal adenocarcinoma.J Mol Diag12:. 433-440.
- 117.Balaguer F, Moreira L.Lozano JJet al(2011) Colorectal cancers with microsatellite instability display unique miRNA profiles.Clin Cancer Res17:. 6239-6249.
- 118.Tuddenham L, Wheeler G.Ntounia-Fousara Set. al(2006) The cartlidge specific microRNA-140 targets histone deaacetylase 4 in mouse cells.FEBS Lett580: 4214-4217.
- 119.Costa Y, Speed R M, Gautier.Pet al(2006) Mouse MAELSTROM: the link between mwiotic silencing of unsynapsed chromatin and microRNA pathways?Hum Mol. Genet15: 2324-2334.
- 121.Saito Y. (2006) Specific activation of microRNA-127 with downregulation of the protooncogene BCL6 by chromatin-modifying drugs in human cancer cells.Cancer. Cell9: 435-443.
- 122.Fazi F, Rosa A.Fatica A et al (2005) A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPalpha regulates human granulopoiesis. , Cell 123, 819-831.
- 123.Boland C R, Thibodeau S N, Hamilton S R, Sidransky D, Eshleman J R et al. (1998) A National Cancer Institute workshop on microsatellite instability for cancer detection and familial predisposition: development of international criteria for the derivation of microsatellite instability in colorectal cancer.Cancer. Res58: 5248-5257.
- 124.Baldas S E, Barta N, Lammers S, Diens H P, Odenthal M. (2005) Label-free analysis of microsatellite instability in colorectal carcinoma by on-chip electrophoresis. Technical Note 5989 2626EN. Agilent Technologies .
- 125.Raptis S, Mrkonjic M, Green R C, Pethe V U, Monga N et al. (2007) MLH1-93 G>A promoter polymorphism and the risk of microsatellite unstable colorectal cancer.J Nat Cancer Inst99:. 463-474.
- 126.Berg K D. (2000) Detection of microsatellite instability by fluorescence multiplex polymerase chain reaction. , J Mol Diag 2, 20-28.
- 127.Banerjea A, Phillips S M, Dorudi S, Bustin S A. (2003) Colorectal cancer with mononucleotide microsatellite instability can be identified using microfabricated chip technology.Anal. Biochem322: 130-133.
- 128.Ahmed F E. (2007) Colon cancer epigenetics: The role of environmental factors & the search for molecular biomarkers.J. , Env. Sci. HealthC25: 75-130.
- 129.Weisenburger D J, Campan M.Long TIet al(2005) Analysis of repetitive element DNA methylation by MethylLifgt.Nucleic Acids Res33:. 6823-6836.
- 130.Eads C A, Lord R V.Wickramasinghe Ket al(2001) Epigenetic patterns in the progression of esophageal adenocarcinoma.Cancer. Res61: 3410-3418.
- 131.Eads C A, Lord R.Kurumboor SKet al(2000) Fields of aberrant CpG Island hypermethylation in Barrett’s esophagus and associated adenocarcinoma.Cancer. Res60: 5021-5026.
- 133.Johnson R A, Wichern D W.(2002)Applied Multivariate Statistical Analysis. Upper Saddle River. , NJ:PrenticeHall
- 134.Tang Y, Ghosal S, Roy A. (2007) Nonparametric Bayesian estimation of positive false discovery rates. , Biometrics 63, 1126-1134.
- 135.Choi H, Nesvizhskii A I. (2008) False discovery rates and related statistical concepts in mass spectrometry-based proteomics.J Proteome Res7:. 47-50.
- 136.. In situ Molecular Pathology and Co-Expression Analysis. Elsevier Science & Tachnology Books,AcademicPress,289 pp Nuovo GJ. (ed.) 2012.
- 137.Petrelli N J, Letourneau R, Weber T, Nava M E, Rodriguez-Bigas M. (1999) Accuracy of biopsy and cytology for the preoperative diagnosis of colorectal adenocarcinoma.J Surg Oncol71:. 46-49.
- 138.Winter M J, Nagtegaal I D, van Krieken JH, Litvinov S V. (2003) The epithelial cell adhesion molecule (Ep-CAM) as a morphoregulatory molecule is a tool in surgical pathology.Am. , J Pathol163: 2139-2148.
- 139.Matsushita H M, Matsumura Y, Moriya Y, Akasu T, Fujita S et al. (2005) A new method for isolating colonocytes from naturally evacuated feces and its clinical application to colorectal cancer diagnosis.Gastroenterology129:. 1918-1927.
- 140.Simpson R J, Lim J E, Moritz R L, Mathivanan S. (2009) Exosomes: proteomic insights and diagnostic potential.Exp Rev roteomics6:. 267-283.
- 141.Kraemer H C.(1992)Evaluating Medical Tests: Objective and Quantitative Guidelines. Sage Publications. , Newsburry Park, California
- 142.Ripley. (1997) Classification. InEncyclopedia of Statistical Sciences. , Wiley-Interscience Publication, NY 1.
- 144.C Y Nagan, Yamamoto H, Seshimo I, Ezumi K, Terayama M et al. (2007) A multivariate analysis of adhesion molecules expression in assessment of colorectal cancer.J Surg Oncol95:. 652-662.
- 145.Reiner A, Yekutieli D, Benjamini Y. (2003) Identyfying differentially expressed genes using false discovery rate controlling procedures.Bioinformatics19:. 368-375.
- 146.Pawitan Y, Michiels S, Kosciely S, Gusnato A, Polner A. (2005) False discovery rate, sensitivity and sample size for microarray studies.Bioinformatics21:. 3017-3024.
- 147.Tang Y, Ghosal S, Roy A. (2007) Nonparametric Bayesian estimation of positive false discovery rates. , Biometrics 63, 1126-1134.
- 148.Choi H, Nesvizhskii A I. (2008) False discovery rates and related statistical concepts in mass spectrometry-based proteomics. , J Proteom Res 7, 47-50.
- 149.Earl-Slatter A (2002) Cross Validation. InThe Handbookmof Clinical Trials and Other Research. , Oxford, UK
- 151.O Y Yildiz.Aslan A and Alpagdin E (2011) Multivariate statistical tests for comparing classification algorithms. Coello C A, ed , Berlin, InLearning and Intelligence Optimization 1-5.
- 152.Hanley J A, McNeil B J. (1982) The meaning and use of the area under a receiver operating characteristic (ROC). 143, 29-36.
- 153.K P Sureh, Chandrashekare S. (2013) Sample size estimation and power analysis for clinical research studies.J Hum Reprod Sci5:. 7-13.
- 154.R G Cornell, Ed. (1984) Statistical models for cancer studies. InModels to Analyze Strategies in the General Population,MarcelDekker,NY. 346-347.
- 156.Wegman E. (1990) Hyperdimensional data analysis using parallel coordinate.J Am Stat Assoc85:. 644-675.
- 158.Willey J C, Crawford E L, Jackson C M, Weaver D A, Hoban J C et al. (1998) Expression measurement of many genes simultaneously by quantitative RT-PCR using standardized mixtures of competitive templates.Am J Resp Cell Mol Biol19:. 6-17.
- 159.DeMuth J P, Jackson C M, Weaver D A, Crawford E L, Durzinsky D S et al. (1998) The gene expression index cmyc x E2F-1/p21 is highly predictive of malignant phenotype in human bronchial epithelial cells.Am J Respir Cell Mol Biol19:. 18-29.
- 160.Lalkhen A G, Mclkey A. (2004) Clinical tests: Sensitivity and specificity. , Crit Care Pain 8, 221-223.
- 161.Chomezynski P, Sacchi N. (1987) Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction.Anal. Biochem162: 156-159.
- 162.El-Hefnawy T, Raja S, Kelly L, igbee W L, Kirkwood J M et al. (2004) Characterization of amplifiable circulating RNA in plasma and its potential as a tool for cancer diagnosis.Clin Chem50:. 564-573.
- 163.Kosaka N, Iguchi H, Ochiyn T. (2010) Circulating microRNA in bodyfluid: a new potential biomarker for cancer diagnosis and prognosis.Cancer. Sci101: 2087-2092.
- 164.Zernecke A, Bizhekov K, Noel H, Shagdarsura E, Gan L et al. (2009) Schoder A and Weber C. Signal2: ra81 .
- 165.Valadi H, Elkstrom K, Bossios A, Sjostrand M, Lee J J et al. (2007) Exosome mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells.Nat Cell Biol9:. 654-659.
Cited by (1)
- 1.Escobar Andres, Diab-Liu Alex, Bosland Kamaya, Xu Chang-qing, 2023, Microfluidic Device-Based Virus Detection and Quantification in Future Diagnostic Research: Lessons from the COVID-19 Pandemic, Biosensors, 13(10), 935, 10.3390/bios13100935