
Abstract
Multiple endocrine neoplasia type 1 (MEN1) is a syndrome emerging from characteristic mutations of MEN1 gene with concurrently enunciated multiple endocrine and tumours and associated non-endocrine neoplasm. Previously designated as Werner’s syndrome, MEN1 syndrome denominates genomic mutation within chromosome 11q13 or a tumour suppressor gene with a distinctive protein product nomenclated as “menin”. MEN1 syndrome demonstrates an autosomal dominant pattern of disease inheritance where genomic mutations delineate a comprehensive (100%) disease penetrance. MEN1 gene was initially identified in 1997 upon chromosome 11q13. Although twelve genetic mutations were primarily identified, currently beyond eighteen hundred genomic mutations are scripted1, 2. MEN1 syndrome is comprised of diverse combination of twenty or more endocrine and non-endocrine tumours exemplifying a classic triad of pituitary, parathyroid and pancreatic neoplasm. Diverse non endocrine tumours enunciated with MEN1 syndrome are denominated with meningioma, ependymoma or angiofibroma1, 2. Endocrine tumours are discerned on account of excessive hormonal secretion engendered from various neoplasm or on account of neoplastic evolution. Approximately 10% instances can occur due to a de-novo genomic variant. Offspring of an individual with MEN1 syndrome quantifies a 50% possibility of inheriting the genomic variant. Cogent prenatal diagnosis can be determined in instances where specific genomic variant of a particular family is known. Physical, psychological and social restrictions are prevalent with MEN1 syndrome. Heterozygotes with MEN1 genetic variant are denominated as carriers and manifest a two- fold possible mortality1, 2.
Author Contributions
Academic Editor: El-Sabbagh AH, professor of plastic surgery, faculty of medicine, mansoura university, Egypt.
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2020 Anubha Bajaj
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Introduction
Disease Characteristics
MEN1 demonstrates variable familial clustering with a prevalence of 1:30,000 to 1 in 500,000 individuals. Majority (90%) of subjects of MEN1 are familial whereas around 10% instances are sporadic. Implicated individuals (100%) delineate a parathyroid adenoma by 50 years of age. Mortality with MEN1 syndrome is enunciated by complications engendered due to pancreatic or gastrointestinal neuroendocrine tumours.
Preponderant chromosomal mutations are family specific whereas three fourths (75%) are inactivating mutations. An estimated 5% to 20% subjects harbour genetic mutations within MEN1 gene encoding region. MEN1 syndrome lacks a genotype- phenotype concurrence, particularly within various enunciated neoplasms1, 2.
Tumour suppressor gene product menin is non-homologous with specific, known proteins although it is structurally conserved across various species such as Drosophila or homo-sapiens. Menin is involved within specific cellular functions such as cellular proliferation, apoptosis and genomic integrity. Nevertheless, non functional menin protein engenders neoplastic transformation through various obscure mechanisms. Multiple endocrine neoplasia type I is indicated in individuals demonstrating the emergence of diverse endocrine tumours although non-endocrine tumours may precede the appearance of hormone secreting endocrine tumours3, 4.
Parathyroid neoplasm are significantly enunciated in endocrinal MEN1 and manifest hypercalcemia or primary hyperparathyroidism with consequent hypersecretion of parathyroid hormone. Primary hyperparathyroidism arising within MEN1 syndrome is commonly a multi-glandular disorder accompanied by enlargement of quadruple parathyroid glands instead of a singular adenoma, thus adoption of imaging modalities may be unsatisfactory for diagnosis. Pituitary neoplasm with MEN1 syndrome is constituted by prolactin secreting anterior pituitary adenoma or prolactinoma which delineates oligo-menorrhoea, amenorrhoea and galactorrhoea in implicated females and sexual dysfunction with infrequent gynecomastia in males. Anterior pituitary adenoma secreting growth hormone demonstrates acromegaly3, 4.
Combined anterior pituitary adenomas secreting growth hormone/ prolactin (GH/PRL) can enunciate acromegaly, oligo-menorrhoea/amenorrhoea/galactorrhoea in females and sexual dysfunction and/or gynecomastia in males. Anterior pituitary neoplasm secreting thyroid stimulating hormone (TSH) exhibit symptoms of hyperthyroidism.
Anterior pituitary adenoma secreting adrenocorticotrophic hormone (ACTH) are predominantly associated with Cushing’s syndrome. Non-endocrine pituitary tumours exemplify neoplastic enlargement with compression of adjacent anatomical structures such as optic chiasma with consequent visual disturbances and/or features of hypopituitarism. Magnetic resonance imaging can appropriately discern particular pituitary neoplasm3, 4.
Well differentiated endocrine neoplasm of gastro-entero- pancreatic (GEP) tract are symbolized by gastrinoma engendering a Zollinger- Ellison syndrome, insulinoma with consequent hypoglycaemia, glucagonoma with emergent hyperglycaemia, anorexia, glossitis, anaemia, diarrhoea, venous thrombosis and cutaneous rash besides a vasoactive intestinal peptide secreting adenoma (VIPoma) enunciating watery diarrhoea, hypokalemia and achlorhydria syndrome. Well differentiated neoplasm of gastro-entero-pancreatic (GEP) tract can delineate tumefaction of the stomach, duodenum, pancreas and intestinal tract4, 5.
Associated clinical presentations are
Zollinger Ellison syndrome (ZES) which manifests as a peptic ulcer in combination with or absence of chronic diarrhoea arising on account of a gastrin- secreting
tumour within duodenal mucosa (gastrinoma).
Hypoglycemia can emerge due to insulin secreting pancreatic tumour (insulinoma).
Hyperglycemia, anorexia, glossitis, anaemia, diarrhoea, venous thrombosis, cutaneous rash (necrolytic migratory erythema) can occur on account of glucagon- secreting pancreatic tumour (glucagonoma).
Watery diarrhoea, hypokalemia, achlorhydria (WDHA syndrome) appears due to vasoactive intestinal peptide (VIP) secreting adenoma (VIPoma).
Adrenocortical tumours exemplify primary hypercortisolism or hyperaldosteronism5, 6. Non- functioning pancreatic endocrine neoplasm which are difficult to discern with cogent biochemical and imaging modalities are frequent in MEN1 syndrome. Type II gastric enterochromaffin -like cell carcinoids are frequently discerned with well differentiated endocrine neoplasm of GEP tract in MEN1. Aforesaid carcinoids usually are incidentally detected during gastric endoscopy performed to diagnose Zollinger Ellison syndrome. Carcinoid tumours are generally non hormone secreting and exemplify an enlarged, organ-specific mass commonly appearing beyond 50 years. Cutaneous manifestations can indicate MEN1 syndrome and usually precede manifestations of endocrinal, hormone secreting neoplasm5, 6.
Endocrine neoplasia exemplified with MEN1 syndrome are distributed as parathyroid (90%), anterior pituitary (30% to 40%), pancreas or gastrointestinal tract (30% to 70%), adrenal cortex and thymus. Non endocrine tumours articulated with MEN1 syndrome are facial skin tumours such as angiofibromas or collagenomas, testicular epididymomas, lipomas, ependymomas, leiomyomas and meningiomas. Non syndromic neoplasm associated with MEN1 demonstrate somatic MEN1 mutations with frequencies such as glucagonoma (60%), vasoactive intestinal peptide adenoma (57%), non functioning pancreatic neuroendocrine tumours (44%), gastrinoma (38%), bronchial carcinoid (35%), parathyroid adenoma (35%), lipoma (28%), insulinoma (2% to 19%), angiofibroma(10%), anterior pituitary tumours (3.5%) and adrenocortical tumour (2%)1, 2. (Table 1, Table 2)
Table 1. Categories of endocrine tumours in MEN1 syndrome1.| Tumour type | Subtype with hormones | Prevalence in MEN1 syndrome |
| Parathyroid | Parathyroid hormone | Primary hyperparathyroidism in 100% by 50 years |
| Anterior pituitary (30%-40% in MEN1) | Secreting prolactinoma | 60% of anterior pituitary tumours |
| Growth hormone secreting | 25% of anterior pituitary tumours | |
| Growth hormone/prolactin secreting | 5% | |
| Thyroid stimulating hormone-secreting | Rare | |
| Adrenocorticotrophic hormone-secreting | <5% | |
| Non-functioning | <5% | |
| Well-differentiated endocrine | Gastrinoma | 40% of well differentiated tumours |
| Insulinoma | <10% | |
| Glucagonoma | <1% | |
| VIPoma | <1% | |
| Non-functioning and pancreatic polypeptide secreting | 20% to 55% of gastro-entero-pancreatic neuro-endocrines | |
| Carcinoid (non-secreting) | Bronchopulmonary | 2% |
| Thymic | 2% | |
| Gastric | 10% | |
| Adrenocortical | Cortisol -secreting | |
| Aldosterone-secreting | 40% | |
| Pheochromocytoma | <1% |
| Tumour type | Differentiation |
| Pituitary neoplasm | Solitary pituitary adenoma not associated with MEN1 and absent findings respond to medical therapy. Multiple pituitary adenoma represent predisposition |
| Zollinger Ellison Syndrome( ZES) | Sporadic tumours of pancreatic origin and appear a decade later. Gastrinomas may appear in MEN4. ZES may concur with RPRD1A and CDKN1B variants |
| Non functioning neuroendocrine tumours (NF-NETs) | Implicate 20% to 55% of MEN1. May concur with VHL and NF1. |
| Insulinoma | Onset a decade later in sporadic insulinoma. NF1 can be associated with Insulinoma |
| Carcinoid tumours | Derivatives of midgut and hindgut, argentaffin reactive, secrete serotonin, benign behaviour when un-associated with MEN1. Coexistent gastric carcinoids and hyper-parathyroidism are indicative of MEN1( not atypical or incomplete). |
| Facial angiofibromas | Discerned in TSC which is delineated at 3 to 4 years (MEN1<40 years) |
| Leiomyomas | Enunciated in Alport’s syndrome |
Clinical Elucidation
Endocrine neoplasm concurring with MEN1 syndrome can delineate a mild form of primary hyperparathyroidism with hypercalcemia emerging in asymptomatic persons or as an initial symptom in a majority (90%) of individuals. Onset of clinical disease is betwixt 20 years to 25 years whereas decline in the bone mass ensues at 35 years and hypercalcemia is apparent by 50 years. Lethargy, depression, altered mental status, declining mental alertness, confusion, obtundation, coma, anorexia, constipation, nausea, vomiting, diuresis, dehydration, hyper-calciuria, renal stones, impaired urinary concentration, enhanced bone resorption, possible emergence of fracture, hypertension and a reduced QT interval on an electrocardiogram are manifestations encountered due to hypercalcaemia engendered with parathyroid neoplasm2, 4.
Hypercalcaemia can enhance gastrin secretion from gastrinoma thus initiating or exacerbating a concurrent Zollinger Ellison syndrome. Post natal hypo-calcemia can ensue. Parathyroid carcinoma is infrequent with MEN1 syndrome.
Pituitary tumours with MEN1 syndrome appear as an initial manifestation in 17% subjects with a variable incidence of 15% to 55%, roughly as 25% of simplex and 10% of familial instances. Pituitary adenoma predominates in females with a female to male ratio of 1.6:1. Pituitary adenomas can produce several hormones such as growth hormone(GH), prolactin(PRL), follicle stimulating hormone(FSH), adrenocorticotropic hormone(ACTH) or luteinizing hormone(LH). Clinical symptoms such as nerve compression, headache or hypopituitarism are significant1, 3.
Frequent, usually solitary prolactin secreting pituitary adenoma or a prolactinoma is associated with amenorrhoea and galactorrhoea in females and decreased libido or impotence in males. ACTH secreting tumours can display features of hypercortisolism. GH secreting adenomas demonstrate gigantism in children and acromegaly in adults5, 6.
Functional FSH secreting pituitary adenomas enunciate a decline in the libido and erectile dysfunction. Majority (65% to 85%) of pituitary adenomas of MEN1 syndrome are enlarged and arise as macro-adenomas. MEN1 associated pituitary adenomas are multiple, especially prolactin and ACTH-secreting adenomas. Although malignant transformation of MEN1 associated pituitary neoplasm is exceptional, an estimated 32% of pituitary macro-adenomas are infiltrative5, 6.
Well- differentiated endocrine neoplasm of gastro- entero- pancreatic tract (GEP) with MEN1 are comprised of gastrinomas in approximately 40% individuals with the enunciation of Zollinger Ellison syndrome demonstrating upper abdominal pain, diarrhoea, oesophageal reflux and peptic ulcer. Hyper-gastrinaemia with undetected Zollinger Ellison syndrome and lack of preceding clinical symptoms can manifest a gastric ulcer with perforation. Heartburn, weight loss and hyper-gastrinaemia secondary to Zollinger Ellison syndrome engenders multiple duodenal ulcers. Classic epigastric pain appears at night following meals within two or more hours. Pain can ensue within right upper quadrant, chest or dorsal trunk. Vomiting occurs due to partial or complete gastric outlet obstruction whereas hematemesis and melena is engendered due to gastro-intestinal bleed. Zollinger Ellison syndrome usually arises prior to 40 years and an estimated 25% instances of MEN1 with Zollinger Ellison syndrome lack a specific family history4, 6.
Endocrine pancreatic micro-adenomatosis is a manifestation of MEN1 syndrome. Characteristically, multiple, miniature gastrinomas beneath < 1 centimetre diameter are observed within the duodenal submucosa. Majority (> 80%) of gastrinomas with MEN1 syndrome are situated within first and second part of duodenum. Duodenal gastrinomas with MEN1 syndrome demonstrate diffuse hyperplasia of gastrin-secreting cells and configure gastrin- producing, multifocal micro-adenomas beneath < 1 millimetre magnitude. An estimated 50% of duodenal micro-gastrinomas depict loss of heterozygosity within MEN1 genetic locus, thereby articulating an initial representation of the neoplastic syndrome6, 7.
Gastrinomas emerging with MEN1 syndrome are frequently multiple and malignant. Almost half (50%) of aforesaid gastrinomas metastasize prior to discernment. Hepatic metastasis is accompanied by an inferior prognosis, in contrast to lymph node metastasis which may not decimate prognostic outcomes. In contrast to MEN1 associated duodenal gastrinomas, aggressive, infrequent pancreatic gastrinomas are enlarged and depict an enhanced incidence of hepatic metastasis 6, 7.
Benign insulinomas associated with MEN1 syndrome generally appear a decade prior to sporadic lesions. Usually a singular tumour is articulated within multiple, islet cell macro-adenomas. The hyper-insulinism engendering neoplasm is usually 1 centimetre to 4 centimetre in dimension. Glucagonomas are infrequent although accompany adjunctive neoplasm appearing in MEN1 syndrome. MEN1 associated glucagonoma comprise of around 3% of glucagon-secreting neoplasm, usually exceed > 3 centimetre in magnitude and demonstrate frequent visceral metastasis. Nearly 80% of MEN1 associated glucagonomas exhibit malignant transformation with hepatic metastasis6, 7.
Vasoactive intestinal peptide secreting adenoma associated with MEN1 syndrome are enunciated in roughly 17% individuals while an estimated 5% are discernible and exceed > 3 centimetre in diameter. Aforesaid adenomas are preponderantly malignant and frequent, discernible liver metastasis 6, 7.
Non-secreting tumours of gastro-entero-pancreatic (GEP) tract are cogitated in an estimated 55% of candidates with MEN1 syndrome and are detectable with ultrasonography. Disease penetrance at 50 years is around 34%. Average life expectancy of individuals with non-secreting GEP neoplasm is reduced, in contrast to subjects devoid of a pancreatico-duodenal tumour.
Carcinoid tumours appearing with MEN1 syndrome are comprised of thymic, bronchial and type II gastric enterochromaffin- like (ECL) carcinoids emerging in around 3% individuals. Thymic carcinoids predominate in males with a male to female proportion of 20:1. Bronchial carcinoids are predominant in females with a female to male ratio of 4:15, 7.
Although carcinoid tumour is aggressive and recalcitrant to therapy, it is accompanied by a gradual clinical course. Thymic, bronchial or gastric carcinoids infrequently hyper-secrete adrenocorticotrophic hormones, calcitonin, growth hormone releasing hormone, serotonin or histamine and can exceptionally delineate carcinoid syndrome.
Thymic carcinoids are aggressive neoplasms and commonly arise in male smokers. Thymic carcinoids can produce growth hormone and engender acromegaly or adrenocorticotrophic hormone and generate Cushing’s syndrome. Median survival of an individual with thymic carcinoid is approximately 9.5 years5, 7.
Gastric carcinoids are discerned at a mean age of 50 years. Gastric carcinoids are detected incidentally during endoscopic procedures in around 70% subjects with MEN1 syndrome and can emerge as multifocal and synchronous or appear disparately.
Bronchial carcinoids arise as multifocal, synchronous tumours or occur within a diverse time period. Adrenocortical neoplasia incriminating a singular or bilateral adrenal glands are discernible by computerized tomography (CT) in approximately 20% to 73% individuals. Concurrent neoplasia are comprised of a variety of cortical adenomas, hyperplasia, multiple adenomas, nodular hyperplasia, cysts or carcinomas with beneath < 10% tumours demonstrating hormonal hypersecretion. Cushing’s syndrome is frequent whereas primary hyper-aldosteronism or primary hyper-cortisolism is infrequent7, 8.
Asymptomatic adrenal gland enlargement can emerge as a hyperplastic process with median tumour magnitude of 3 centimetres although majority of neoplasms are beneath <3 centimetres. Comprehensive incidence of adrenocortical carcinoma is approximately 1%. MEN1 syndrome demonstrating adrenal neoplasm exceeding 1 centimetre magnitude are accompanied by probable malignant emergence in around 13% subjects and the possibility is augmented in neoplasia exceeding >4 centimetres6, 8.
Non endocrine tumours associated with MEN1 syndrome usually display cutaneous lesions in around 85% individuals as with facial angiofibromas which are benign neoplasia composed of blood vessels and connective tissue.
Non- regressive acneiform papules extending beyond vermilion border of the lips can appear. Collagenomas emerge in roughly 70% individuals and configure multiple, skin coloured, hypo-pigmented cutaneous nodules symmetrically articulated upon the trunk, neck and upper limbs. Nodules are typically asymptomatic, spherical, firm, elastic and vary in magnitude from several millimetres to centimetres. Lipomas are universally situated in subcutaneous tissue or visceral locales and discerned in around 30% subjects7, 8.
Café au lait macules are articulated in nearly 38% individuals whereas confetti like hypo-pigmented macules and multiple gingival papules appear in approximately 6% persons.
Tumours of central nervous system (CNS) are infrequent with MEN1 syndrome. Meningiomas appear in nearly 8% individuals, are essentially asymptomatic and around 60% are devoid of further expansion. Ependymomas arise in almost 1% subjects7, 8. Leiomyomas as benign neoplasia which appear to be derivatives of smooth (non striated) muscle. Sporadic leiomyomas are enunciated in nearly 20% to 30% females of reproductive age group. Thyroid neoplasia exemplified in MEN1 syndrome are comprised of adenomas, colloid goitre or thyroid carcinoma and appear in exceeding> 25% individuals6, 8.
Figure 1, Figure 2, Figure 3, Figure 4, Figure 5, Figure 6, Figure 7, Figure 8
Figure 1.MEN1 syndrome demonstrating various components and neoplasm10.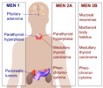
Figure 2.MEN1 syndrome delineating autosomal pattern of inheritance with possible modes of transmission11.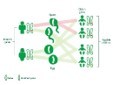
Figure 3.MEN1 syndrome with proportionate distribution of classical neoplasm12.
Figure 4.MEN1 syndrome demonstrating a parathyroid adenoma with encapsulation and glandular proliferation13.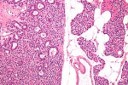
Figure 5.MEN1 syndrome demonstrating goitrous enlargement of thyroid gland with colloid impaction13.
Figure 6.MEN1 syndrome designating a pancreatic polypetide secreting tumour with benign appearing adenomatous glands14.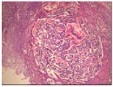
Figure 7.MEN1 syndrome denominating a pancreatic neuroendocrine neoplasm with accumulation of benign, adenomatous glands15.
Figure 8.MEN1 syndrome enunciating an adrenocortical carcinoma with features of malignancy such as nuclear hyperplasia, hyperchromasia and pleomorphism16.
Disease Supervision
Surveillance for MEN1 syndrome is life long and is commenced in early childhood. Preliminary detection and therapeutic intervention of potentially malignant neuroendocrine neoplasia can decimate the morbidity and mortality associated with MEN1 syndrome.
In order to competently evaluate emergence of multiple endocrine neoplasia type 1 (MEN1) certain investigations are recommended. Annual biochemical investigations comprise of assessment of serum prolactin, insulin-like growth factor (IGF-1), fasting glucose and insulin commencing at 5 years. Fasting total serum calcium and ionized serum calcium, chromogranin A, pancreatic polypeptide, glucagon and vasoactive intestinal peptide (VIP) for evaluatingadjunctive pancreatic neuroendocrine tumours (NET) require initiation at 8 years1, 3.
Fasting serum gastrin values are assessed at 20 years, especially in individuals demonstrating gastro-oesophageal reflux or diarrhoea. Assay of fasting serum intact parathyroid hormone (PTH) is beneficial. Obscure genetic status of MEN1 syndrome can delineate 50% possible emergence and can be categorized with aforesaid investigations.
At three to five year interval, imaging of cogent neoplasia related to MEN1 syndrome is mandated, contingent to concurrent biochemical indications of a particular neoplasm along with or without the symptoms of an MEN1 related tumour(1,3). Screening and surveillance is necessitated in specific, kindred subjects. Insulinoma and pituitary adenoma commonly appears at 5 years, parathyroid adenoma usually arises at 8 years, gastrinoma generally emerges at 20 years, adrenal gland is implicated before < 10 years and thymic tumours arise roughly prior to 15 years of age2, 3.
Computerized tomography (CT) and magnetic resonance imaging (MRI) of the abdomen can be initiated at 20 years of age whereas a cranial MRI is required at 5 years. Annual computerized tomography of the chest can be beneficial.
Somatostatin receptor scintigraphy (SRS) octreotide scan can also be adopted on an annual basis. Fasting serum parathyroid hormone (PTH) values and an annual chest CT is advantageous2, 4. Probable emergence of MEN1 in blood relatives necessitates cogent evaluation. As preliminary tumour discernment is beneficial and influences therapeutic management, molecular genetic testing can be adopted to evaluate pertinent family members2, 4.
Diagnostic Manifestations
With variable prevalence of MEN1 syndrome, disease penetrance is 50% within 20 years and almost comprehensive (95%) within 40 years. Criterion for characterizing MEN1 syndrome are constituted of concurrent neoplasm arising within two of three endocrine organs as cogitated with parathyroid, pituitary or well differentiated endocrine tumours of gastro- entero- pancreatic tract with an associated enlarged tumefaction or hyper-secretion of polypeptide hormones6, 8.
Familial MEN1 syndrome is characterized within an individual demonstrating –
minimally, a singular first degree relative with one or more of aforesaid endocrine tumours.
incrimination of a singular organ in combination with an MEN1 genomic variant.
Classical MEN1 syndrome is designated as the occurrence of endocrine tumours exceeding >1, which can be of parathyroid, pituitary or gastro-entero-pancreatic (GEP) tract origin. Biochemical evaluation demonstrates enhanced serum levels of parathyroid hormone and calcium in primary hyperthyroidism, enhanced serum prolactin levels in a prolactinoma, enhanced serum values of gastrin, insulin or vasoactive intestinal peptide with tumours of gastro-entero-pancreatic tract6, 8.
Clinical manifestations, antecedent disease discernment and suitable treatment of metabolic manifestations can safely exclude complications of primary hyperparathyroidism or concurrent Zollinger Ellison syndrome, conditions which engender mortality in MEN1 syndrome.
Malignant conversion in MEN1 syndrome account for nearly 30% mortalities. Individuals with MEN1 syndrome can enunciate premature mortality. Minimal intra-familial inheritance is denominated with pituitary adenoma, adrenal neoplasm and thymic neoplasm6, 8.
Investigative Assay
Endoscopic ultrasound (EUS) is a sensitive imaging methodology for delineating miniature, below < 10 millimetre magnitude pancreatic endocrine tumours arising in an asymptomatic individual with MEN1. Pancreatic gastrinoma can be adequately evaluated with computerized tomography (CT), magnetic resonance imaging (MRI) and /or an endoscopic ultrasound (EUS)1, 2.
A cogent magnetic resonance imaging (MRI) can be adopted to discern prolactinoma. Somatostatin receptor scintigraphy (SRS) can be employed to detect neuroendocrine tumours (NET) and endoscopic ultrasound is applicable in the delineation of pancreatic endocrine tumours.
Molecular genetic testing of MEN1 is capable of distinguishing heterozygous MEN1 gene in a majority (80% to 90%) subjects with familial MEN1 syndrome and in around three fifths (65%) of individuals with simplex MEN1 syndrome within the family1, 2. Emergence of multiple endocrine neoplasia type 1 (MEN1) can be established with
Two of three endocrine organs demonstrating neoplasia as cogitated with parathyroid, pituitary and well differentiated endocrine tumours of gastro-entero- pancreatic (GEP) tract.
Occurrence of heterozygous genetic variant within MEN1 syndrome as denominated upon molecular analysis.
Molecular assay can incorporate evaluation of a singular gene or employment of a multigene panel with comprehensive genomic testing. Singular genetic testing with sequence analysis of genome indicative of MEN1 is initially performed followed by a gene-specific deletion /duplication/ analysis1, 2. Multi-genetic panel assay which enunciates chromosomal mutation of MEN1 syndromic and associated genes can be employed. Certain genes are of uncertain significance and may lack concurrence with specific phenotypes. Methodologies of molecular analysis are comprised of sequence analysis, genomic deletion/ duplication with or without non- sequencing based assays. Extensive genetic testing can be accomplished with exome sequencing or genome sequencing which may indicate previously unconsidered diagnoses. Chromosomal mutations of diverse or various genes can incur an identical clinical representation1, 2.
Recognition of MEN1 syndrome associated genomic mutation can be assessed with polymerase chain reaction (PCR) induced amplification of exons and splicing sites. Aforesaid manoeuver, however, is unable to identify subsets of MEN1 syndrome with an absence of genetic mutation within the encoding region. Adoption of next generation sequencing circumvents diagnostic limitations of diverse methodologies. Mutational analysis can be adopted in
asymptomatic, first degree relatives of established MEN1 syndrome
subjects with minimally two classical tumours associated with MEN1 syndrome
a singular MEN1 associated tumour with a cogent family history of MEN1 syndrome
subjects with early onset of MEN1 syndrome below < 30 years with characteristic MEN1 manifestations such as hyperparathyroidism or Zollinger Ellison syndrome1, 2.
Assessment of postoperative hypoparathyroidism requires an assay of serum parathyroid hormone (PTH) values evaluated on day one following subtotal or total parathyroidectomy, which are a superior indicator of residual parathyroid function. Repetitive assessment of serum calcium is an advantageous and cost effective manoeuver. On radiological assessment, features such as osteoporosis, terminal tuft erosion, distal phalanges and nephrolithiasis are exemplified in subjects delineating parathyroid adenoma and hypercalcemia1, 2. Calvarial thickening, frontal bossing, sinus enlargement (frontal sinus), enlargement of sella turcica, prognathic mandible and enhanced incidence of vertebral fractures can be discerned in subjects with acromegaly due to pituitary adenoma.
Computerized tomography(CT) and magnetic resonance imaging (MRI) adequately discerns thymic carcinoid tumour whereas plain chest radiography and somatostatin receptor scintigraphy( SRS) enunciates minimal sensitivity of detection3, 4.
Assessment of urinary catecholamines is mandated prior to surgical intervention in MEN1 syndrome to discern and manage a pheochromocytoma in order to prevent potential hypertensive crisis1.
Therapeutic Options
MEN1 syndrome requires evaluation within specific categories of multi-glandular parathyroid disorder, prolactinoma, gastrinoma and associated gastro-entero- pancreatic neuroendocrine neoplasia in addition to a comprehensive genetic consultation. Primary hyperparathyroidism is preferentially treated with subtotal parathyroidectomy along with cryopreservation of parathyroid tissue or a total parathyroidectomy with auto-transplantation of parathyroid tissue, procedures which can be employed for managing extensive disease or repetitive, therapeutic surgical intervention8, 9.
Calci-mimetics are advantageously administered in primary hyperparathyroidism where a surgical intervention is contraindicated or inadequate. Bone anti-resorption agents are adopted preceding to surgical therapy in order to reduce hypercalcemia and restrict bone resorption.
MEN1 associated pituitary tumours generally necessitate aggressive therapeutic management, in contrast to sporadically emerging neoplasm8, 9. Pituitary adenomas especially prolactinomas can be managed with administration of dopamine agonists such as cabergoline, bromocriptine, pergolide or quinagolide. Cabergoline has minimal side effects and is considered efficacious.
Surgical resection of neuroendocrine tumours can be advantageous. Minimally invasive trans-sphenoidal surgery or radiotherapy is employed for treating recalcitrant, drug- resistant neoplasms and macro-adenomas compressing abutting anatomical structures with consequent neuro- ophthalmological complications8, 9.
Non-secreting pituitary adenoma is preferably managed with a cogent, trans-sphenoidal, surgical extermination. Enlarged adenomas can be accessed with a trans--frontal incision. Approximately 5% to 15% instances are administered potent dopaminergic agonists or somatostatin analogues in order to shrivel the neoplasm prior to a surgical intervention. Growth hormone secreting tumours engendering acromegaly are appropriately managed with trans-sphenoidal surgery which is efficacious in nearly 50% to 70% instances. Somatostatin analogues such as octreotide or lanreotide can be adopted to medically manage growth hormone secreting tumours. Dopamine agonists are beneficial in treating mixed growth hormone-prolactin (GH-PRL) secreting adenomas, which can be administered in around 10% to 20% of neoplasms refractory to somatostatin analogues. Adrenocorticotrophic hormone (ACTH) secreting tumours are managed with surgical excision of the adenoma. ACTH secreting tumours engendering Cushing’s syndrome can be subjected to radiotherapy to decimate hormonal production8, 9.
Well differentiated tumours of gastro-entero-pancreatic (GEP) tract with frequently enunciated gastrinoma are medically treated with proton pump inhibitors and histamine 2 (H2)- receptor blockers in order to decimate the production of gastric acid. Surgical eradication of gastrinoma in MEN1 syndrome is debatable as microscopic neoplasia are disseminated upon entire neuroendocrine tissue, thereby exemplifying an unsatisfactory surgical outcome. Nevertheless, pre-operative imaging, intra-operative exploration and pathologic assessment of surgical specimens of the duodenum and first jejunal loop are essential diagnostic and therapeutic manoeuvers8, 9.
Pancreatic neoplasm associated with MEN1 syndrome are comprised of insulinoma or adjunctive tumefaction and are suitably manged with a surgical eradication. Advanced, metastatic malignancies unamenable to surgical extermination are managed with administered somatostatin analogues, cytotoxic chemotherapy, inhibitors of tyrosine- kinase receptors such as sunitinib or inhibitors of mammalian target of rapamycin(m TOR) as with everolimus. Employment of aforesaid medications enhances median progression free survival8, 9.
Non functioning pancreatic neuroendocrine (NET) tumours are subjected to surgical resection when exceeding > 1centimetre or > 2 centimetre magnitude. Occult tumour metastasis appears as an initial disease representation in a significant proportion of instances. Carcinoid tumours are suitably managed with long acting somatostatin analogues which curb hormonal hyper-secretion accompanying the neoplasm. Surgical eradication of carcinoid tumour is a preferred therapeutic option8, 9.
Metastatic disease or neoplasm unamenable to surgery can be treated with radiotherapy or cogent chemotherapeutic agents. Appropriate therapeutic management of adrenocortical tumours lacks consensus guidelines. Malignant conversion is enhanced in tumours exceeding > 4 centimetre diameter. Surgical extermination is thus indicated for neoplasm exceeding > 4 centimetres, betwixt 1 centimetre to 4 centimetres and neoplasm depicting atypical or debatable radiological features or neoplasm delineating significant growth within preceding 6 months. Besides, surgical eradication of adrenocortical tumours exceeding 3 centimetre magnitude can circumvent a possible malignant conversion8, 9.
Possibility of malignant transformation is enhanced with tumours of duodenum, pancreas or lungs (bronchial carcinoids). Therefore, ablative surgery is not beneficial in treating tumours of aforesaid organs. Comprehensive, prophylactic thymectomy can circumvent the occurrence of a thymic carcinoid, particularly in male smokers. Well differentiated gastro-entero-pancreatic (GEP) tract neoplasia can be managed with administration of somatostatin analogues which essentially curtail proliferation of enterochromaffin like cells. Additionally, peptide receptor radionuclide therapy (PRRT) along with application of radiolabelled somatostatin analogues (SSA) is beneficial in treating MEN1 syndrome associated neoplasia. Aforesaid agents adopt specificity of analogues for somatostatin receptors in order to provide cytotoxic quantities of radioactive isotopes such as yttrium-90 or luteticium -177, particularly in gastro-entero-pancreatic neuroendocrine tumour (GEP-NET) cells8, 9.
Targeted molecular therapies are advantageous and contemporary in treating tumours of MEN1 syndrome. Everolimus is an agent employed as an m TOR pathway inhibitor and is adopted in managing advanced, low grade or intermediate grade pancreatic neuroendocrine tumours (p NETs). As an agent, m TOR regulates cellular survival, proliferation and mobility. Subjects treated with m TOR pathway inhibitors demonstrate an increased median free survival8, 9. Sunitinib is an oral tyrosine- kinase inhibitor which targets vascular endothelial growth factor (VEGF) receptor. Sunitinib is employed for treating advanced pancreatic neuroendocrine tumours (p NETs) enunciating enhanced quantities of vascular endothelial growth factor (VEGF) receptors8, 9.
References
- 1.Giusti F, Marini F. (2017) Multiple endocrine neoplasia type 1 (MEN1)” Gene Reviews: Univ of Washington. , Seattle
- 2.Caplin M E, Pavel M. (2014) Lanreotide in metastatic entero-pancreatic neuroendocrine tumours”. , N Engl J Med: 371, 224-33.
- 3.Carroll R W. (2013) Multiple endocrine neoplasia type 1 (MEN1). , Asia Pacific J Clin Oncol: 9, 297-309.
- 4.Concolino P, Costella A. (2016) Multiple endocrine neoplasia type 1 (MEN1) : an update of 208 new germline variants reported in the last nine years”. , Cancer Genet : 209, 36-41.
- 5.Daglar H K, Kirbas A. (2016) Management of a multiple endocrine neoplasia type 1 during pregnancy : a case report and review of literature”. , J Exp Ther Oncol : 11, 217-20.
- 6.Diaz- Soto G. (2013) Linglart A et al” Primary hyperparathyroidism in pregnancy “ Endocrine. 44, 591-7.
- 7.Falconi M, Erikkson B. (2016) ENETS consensus guidelines update for the management of patients with functional pancreatic neuro-endocrine and non- functional pancreatic neuroendocrine tumours”. , Neuroendocrinology: 103, 153-71.
- 8.Giusti F, Cianferotti L. (2016) Cinacalcet therapy in patients affected by primary hyperparathyroidism associated with multiple endocrine neoplasia syndrome type 1 (MEN1)”. , Endocrine: 52, 495-506.