
Abstract
Background
Overuse of beta-lactam antibiotics has lead to selection for extended-spectrum β-lactamase (ESBL) producing Enterobacteriaceae, a major cause of antibiotic resistant urinary tract infections (UTIs). Standard detection methods are time-consuming, with disputed accuracy.
This study describes a novel real-time PCR method to detect CTX-M, SHV, OXA and TEM.
Methods
179 Enterobacteriaceae isolates from UTIs were collected from the Leicester Royal Infirmary, UK. A multiplex Plexor®-based real-time PCR assay detected ESBLs using their specific amplicon melting temperature, during each cycle, removing the need for a melt-curve analysis. Validation was achieved by end-point PCR and disk diffusion.
Results
The method was able to produce rapid and accurate results, achieving a sensitivity and specificity of 94.9% and 72% respectively, and the assay can differentiate between the different ESBL genes, with ease.
Conclusions
With further investigation, a Plexor®-based assay could form the basis of a high-throughput kit that health services could use to detect ESBLs or other antibiotic resistance genes.
Author Contributions
Academic Editor: Jianping Pan, Department of Clinical Medicine, Zhejiang University City College School of Medicine, China.
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2018 Ruth Reid, et al.
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Introduction
Antibiotic resistance is a natural process, whereby bacteria exchange genes via horizontal transfer, however the overuse of antibiotics has caused a permanent selective force for resistance mechanisms. 1 In the O’Neill report in 2016, it was estimated that every year at least 700,000 people die as a result of antibiotic resistant infections. 2
Extended-spectrum β-lactamases (ESBLs) are one of the most common types of antibiotic resistance found in Enterobacteriaceae and are a major public health concern, both in hospital settings and in the community. 3, 4 First discovered in the 1980’s, ESBLs have since increased massively, becoming a major public concern. 1
ESBLs are able to hydrolyze extended - spectrum cephalosporins (cefotaxime, ceftriaxone, ceftazidime and cefepime) and monobactams (aztreonam). 4, 5 The most common families of ESBLs found are CTX-M, SHV, OXA and TEM. 1
Urinary tract infections (UTIs) are the most common infection found in humans. 6The majority of both community-acquired and nosocomial UTIs are caused by uropathogenic Escherichia coli (UPEC) strains. 6 Serious infection by ESBL-producing UPEC can be linked to increased length of hospital stay, admittance to an intensive care unit, catheterization, increased mortality, morbidity and healthcare cost. 7, 4, 8, 9 Therefore, early detection would benefit both the patient and the health care provider. Screening for ESBL-producing uropathogens is commonly achieved by means of selective agar plates, such as the commercially available ESBL agar. Isolates are also tested for antibiotic sensitivity by disk diffusion or broth microdilution. 5 These procedures can take 4-6 days to obtain an antibiotic resistance profile after the culture, isolation and characterisation of pathogens from a patient sample.
In the O’Neill report, it was suggested that rapid diagnosis could change the way we use antibiotics. Reducing the time spent on broad-spectrum antibiotics by prescribing a more appropriate treatment, increases the favorability of the outcome of the patient and shortens the stay in hospital. Rapid detection of ESBL isolates also has other benefits, including infection control and prevention of outbreaks through the use of hospital hygiene precaution measures. 7, 10, 11 Therefore, rapid and sensitive detection of infections thought to be of the ESBL type is of the utmost importance for patient outcome, the economy and outbreak control.
In this study, a multiplex real-time assay was developed and validated to detect the most prevalent ESBL-producing genes in bacterial isolates found in UTIs, based on the amplicon melting temperature.
Methods
Bacterial Isolates and DNA Extraction
Bacterial isolates (n=181) of Enterobacteriaceae isolated from urinary tract infections were obtained from the Leicester Royal Infirmary hospital (Leicester, England). The Leicester Royal Infirmary was chosen as a collection site as a large number of samples are received from all over the Midlands, giving a wide population base. 25 of the samples were non β-lactamase producing strains of Enterobacteriaceae, and 156 were β-lactamase producing strains. The presence or absence of ESBL production was previously identified by disk diffusion. We collected waste material (agar plates) and therefore informed consent from patients was not required. Both catheter and non catheter patients, symptomatic and non symptomatic patients were included. Four control isolates were incorporated: NCTC 13353 (CTX-M-15), NCTC 13351 (TEM-3), NCTC 13368 (SHV-18) and NCTC 13442 (OXA-48), obtained from Public Health England. Each isolate was sub-cultured on Müller-Hinton agar plates for 24 hr. at 37°C. 2-3 colonies were picked and suspended in 50 µl sterile distilled water and then heated in a AccuBlock dry bath (Labnet International Inc,Windsor, Berkshire) for 10 min at 94°C.
Primers
RT-PCR
The Plexor® qPCR (Promega,Southampton, UK) mastermix kit was used for its suitability in multiplex real-time PCR and its previous use in diagnostic assays. The Plexor® qPCR system mechanism has been previously described. 13 In typical real-time PCR assays, the accumulation of a product is accompanied by an increase in fluorescence. However, the Plexor® qPCR system differs in that the accumulation of a product leads to a decrease in fluorescence. Primers are easy to design, and less expensive than using probes. Primer design requires the addition of an iso-dC label adjacent to a fluorescent reporter on one strand. The Plexor® mastermix contains a dabcyl-iso-dGTP label as a quencher. This can be seen in Figure 1. Plexor® primers are less expensive than probes, and have a higher specificity than SYBR green chemistry. 13
Figure 1.The Plexor® qPCR requires a florescent reporter, adjacent to an Iso-label, to be attached to one of the primers in each pair. Florescence is then quenched by incorporation of a Dabcyl-iso-dGTP label contained in the mastermix, when bound to another DNA strand during the extension phase of the PCR cycle. Therefore , as a product increases , florescence decreases. When the amplicon melts, the strands dissociate and the quencher is released. Shorter amplicons melt at lower temperature to longer ones, allowing differentiation between the product.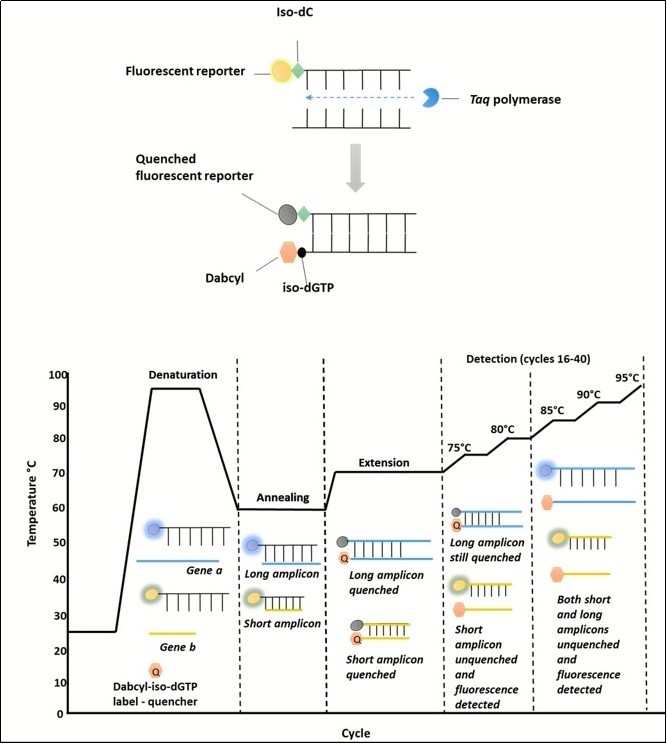
RT-PCR conditions such as annealing temperature and primer concentration were optimised in simplex, and once satisfied, in multiplex. The amplicons of the control isolates were initially tested using high resolution melting curve analysis, first in simplex and then multiplex to determine the melting point of the amplicon. An increase in temperature increments between 84°C and 86°C was implemented to aid resolution between the amplicon melting points of CTX-M and TEM.
Assays were performed in 12.5 µl reactions consisting of 6.25 µl mastermix, 0.5 µl of each primer mix, (forward and reverse primer combined) at a final concentration of 200 nM and 2.5 µl template DNA. The five previously mentioned controls were included in the assay, along with a no template control (NTC).
The RT-PCR amplification protocol was performed in a PikoReal® 96 well RT-PCR platform (ThermoFisher, Loughborough, UK). The RT-PCR amplification program comprised of the following steps: initial denaturation for 10 min at 95 °C, and then 40 cycles of 95 °C for 10 s, annealing at 70°C for 40 s (gradual temperature decrements of 0.2°C per cycle, final annealing temperature 60°C), and fluorescence readings were taken from cycle 16 onwards at 72°C, 76°C , 78°C, 84°C, 84.2°C, 84.4°C, 84.6°C, 84.8°C, 85°C, 85.2°C, 85.4°C, 85.6°C, 85.8°C, 86°C, 88°C and 90°C for 10 s each.
Limit of Detection
For each of the ESBL types, the limit of detection was determined in triplicate with five 1:10 dilutions using DNA extracted by the method mentioned previously. DNA concentration was measured using a Qubit 3 fluorometer.
Validation
ESBL production was confirmed phenotypically via the MAST ID double disc synergy method outlined by the British Society of Antimicrobial Chemotherapy (BSAC). Discs contained Cefotaxime, Ceftazidime and Cefpodoxime with their clavulanic acid counterpart. Plates were incubated for 18 hours at 37°C. A zone of inhibition difference of 5mm or more indicated a positive result for ESBL production.
A multiplex end-point PCR assay was used as a reference for validation. PCR amplification reactions were performed in a volume of 25μl containing 5μl buffer, 2μl mgcl2, 0.5 μlDNTPs, 0.125μl taq polymerase, 400 nM concentrations of each primer, and 2.5μl of DNA template. Cycling parameters for the multiplex assay were as follows: an initial denaturation at 94°C for 5 min; followed by 35 cycles of 94°C for 30 s, annealing at 65°C for 1 min (gradual temperature decrements of 0.5°C per cycle, final annealing temperature 48°C), and 72°C for 1 min; and with a final extension at 72°C for 10 min. Primers can be found in Table 2.
Table 2. Primers used in the validation end-point PCR assay.| Primer | Forward Primer | Reverse Primer | References |
| TEM-1 | CGG ATG GCA TGA CAG TAA GAG | AGG ACC ACT TCT GCG CTC G | Own study |
| SHV-18 | CTCAAGGATGTATTGTGGTTATGC | CTA CGA GCC GGA TAA CGC G | Own study |
| CTX-M | CGTCATCTATGTTCGCCGAC | GCATCTCAGTCGGATCGAGC | Own study |
| OXA-48 | CGGAATGCCTGCGGTAGCAAAG | CAGCCCTAAACCATCCGATG | Own study |
Statistical Analysis
The sensitivity, specificity, positive likelihood ratio and negative likelihood ratio was calculated using SPSS version 22. 95% confidence intervals for the sensitivity and specificity were calculated using exact Clopper-Pearson confidence intervals. 14 Whereas the Log method was used to calculate 95% confidence intervals for the likelihood ratios. 15
Results
A Plexor®-based multiplex real-time PCR assay was designed to simultaneously detect the ESBL genes CTX-M, TEM, OXA and SHV, using amplicon melting temperatures, without the use of melting curve analysis. Figure 2 shows the results of the development of the real-time PCR method Table 1.
Table 1. Primers used in this study.| Oligo | Forward | Reverse | Reference |
| OXA 48 | AGCAAAGGAATGGCAAGAAA | CGCCCTGTGATTTATGTTCA | 12 |
| SHV | GGTCAGCGAAAAACAYCTTG | GCCTCATTCAGTTCCGTTTC | 12 |
| TEM | GATACGGGAGGGCTTACCAT | GGATGGAGGCGGATAAAGTT | 12 |
| CTX-M | AATCTGACGCTGGGTAAAG | CCGCTGCCGGTTTTATC | 12 |
Figure 2.Data showing the change in fluorescence at cycle 16-31 (when florescence) at low temperature and at the meeting of each amplicon. Data was normalized to the highest relative fluorescent unit (RFU). At temperature below the Tm, fluorescence decreases as product Increases each cycle. This is due to the Incorporation of the Dabcyl-iso-dGTP quencher contained within the mastermix. At higher temperatures, fluorescence is very low as the majority of fluorescent reporters have been quenched. This change in fluorescence occurs at different temperatures for each amplicon, therefore, the different products can be distinguished based on temperature.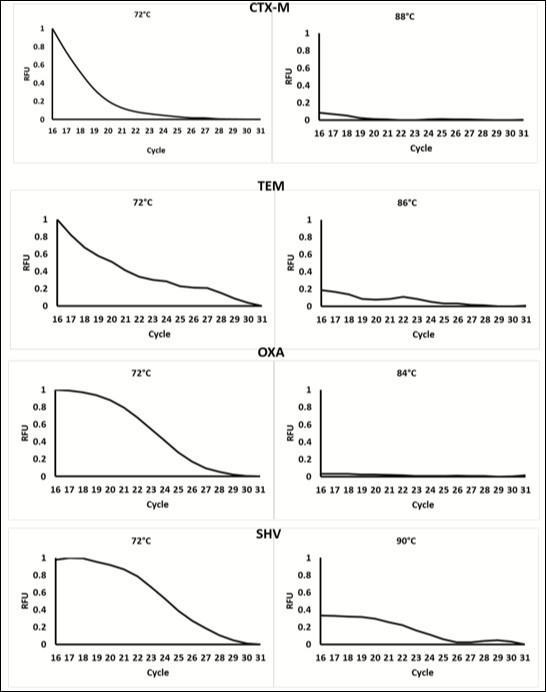
179 samples were collected from the Leicester Royal Infirmary and tested by RT-PCR. The result of the multiplex assay were mostly in agreement with the results of the multiplex end-point PCR. The assay correctly identified 91.7% isolates tested. A summary of the statistical results can be found in Table 3. Seven very major errors and seven major errors were identified. Very major errors were defined as samples that showed as negative for ESBL genes by the RT-PCR assay, but positive by end-point PCR. Major errors were defined as samples that showed as positive for ESBL genes by the RT-PCR assay, but negative by end-point PCR.
Table 3. Summary of statistical analysis.| RT-PCR | Disk Diffusion | |
| Accuracy | 91.71% (86.70-95.29) | 87.36% (81.64 – 91.82) |
| Sensitivity | 94.87% (90.15-99.97) | 96.82% (92.72 – 98.96) |
| Specificity | 72% (50.61-87.93) | 28.00% (12.07 – 49.39) |
| Positive likelihood ratio | 3.39 (1.81-6.36) | 1.34 (1.05 – 1.72) |
| Negative likelihood ratio | 0.07 (0.03-0.15) | 0.11 (0.04 – 0.33) |
Interestingly, the real-time PCR assay picked up additional TEM genes in two of the isolates, which the end-point PCR had failed to. The NTC was assigned correctly as predicted.
In comparison, the disk diffusion method correctly identified ESBL production in 87.36% of tested isolates. Whilst the sensitivity was slightly higher than the RT-PCR, the specificity was significantly lower (see Table 3). Eighteen very major errors and five major errors were identified.
Limit of Detection
The limit of detection for each of the set of primers CTX-M, TEM, OXA and SHV was 0.0004125ng/µl, 0.0242ng/µl, 0.000404ng/µl, and 0.000362 respectively. As TEM was not detected below 0.0242ng/µl, it is recommended that at least this amount of DNA is included in the test.
Discussion
Although other methods have been described for the detection of ESBLs, to our knowledge, this is the first study to accurately and rapidly describe a multiplex real-time PCR assay to detect ESBLs, based on amplicon melting temperatures, without the need for a high-resolution melting curve analysis. Whilst there are other chemistries applied in real-time PCR that do not require high-resolution melt-curve analysis for detection, such as Taqman, these tend to be far more expensive and require a higher level of user experience. 13 In this assay, the detection of products is based upon the melting temperature of amplicons, within each cycle. In the present study, 179 samples previously identified as ESBL-producers by disk diffusion methods, were tested using a Plexor®-based multiplex real-time PCR assay for the detection of 149 variants of the CTX-M, TEM, OXA and SHV genes in one amplification. The assay was able to correctly detect genes in 91.7% of the isolates in under 3 hours. In comparison, disk diffusion methods were able to correctly detect ESBL production in 87.4% of isolates. Whilst in this study, the disk diffusion method showed a slightly higher sensitivity than the RT-PCR method, this has not been seen in other studies. 16, 17 It has also been reported that clinical failures may occur, even when an isolate is negative phenotypically. 18 This may have been seen in this study, as there were eighteen cases where ESBL genes were detected by end-point PCR, but the isolate was negative phenotypically. This further advocates the use of genotypic detection.
ESBL-producing Enterobacteriaceae are a global growing concern, especially when it comes to UTIs. Current susceptibility testing requires at least 24 hr., therefore it is common practice to treat with empirical antibiotic therapy, without full pathogen information. Treating in this way can lead to ineffective therapy, causing an increase in clinical symptoms, including the possibility of urosepsis and ascending infection. It is well known that empirical treatment with antibiotics increases the population of resistant pathogens. 19 Therefore, it is crucial that reliable, accurate and rapid detection methods of antibiotic resistance are available. In addition, prevalence studies are highly important for monitoring the spread and evolution of antibiotic resistance genes.
Not only can the assay detect ESBL production in isolates in under 3 hours, it can also differentiate between the major classes of ESBL, aiding in surveillance studies. In addition, this assay did not require any laborious primer modifications and the results are easy to interpret, with no further analysis required. The results from this assay could lead to treatment escalation much more quickly than current methods. However, due to the low number of negative samples tested in this study, it is difficult to determine if treatment de-escalation could be concluded from the results of this assay. Nevertheless, due to the increased expense of PCR-based methods compared to current susceptibility testing methods, it is likely that only those patients who are suspected of having an ESBL producing UTI, such as patients who suffer from recurrent UTIs or those that are at higher risk (pregnant women, children, immunocompromised) would be tested. Therefore, the patient population tested in this study, represents the samples that would most likely be tested in practice. Until the negative likelihood ratio of this test can be more reliably proven, it is suggested that this test be used as an add-on to current testing in any patients that do not fall into the above criteria.
In the future, more targets could be added to this multiplex assay, as long as they have a different amplicon melting temperature. This is achieved through primer design, as amplicon melting temperature is related to length and the ratio of CG/AT content. As technology improves, the melting temperature difference required between amplicons can decrease, further increasing the number of targets possible in the assay. We recommend that primers for other Carbapenamases and ampCs (other major antibiotic resistance genes) are added, to give this assay full breadth across the antibiotic resistance spectrum seen in UTIs.
A multiplex Plexor®-based real-time PCR assay could provide a rapid, more sensitive, easy to interpret detection method, thereby helping to prevent the inappropriate use of antibiotics. It was found that this method could aid antibiotic susceptibility testing, if ESBL production is suspected. With further investigation this assay could form the basis of a high throughput kit that health services could use to detect ESBLS or other antibiotic resistance genes.
Funding
This work was funded by the De Montfort University Fees Only PhD Scholarship awarded to Ruth Reid.
Acknowledgements
The authors would like to acknowledge Christopher Holmes of University Hospitals of Leicester NHS Trust, Adrian Slater of De Montfort University and Avninder Bhambra of De Montfort University.
References
- 1.Canton R, Gonzalez-Alba J, Galan J. (2012) CTX-M enzymes: origin and diffusion. , Front Microbiol 3.
- 2.O’Neill J. (2016) Tackling drug-resistant infections globally: Final report and recommendations. The review on antimicrobial resistance. HM Government and The Wellcome Trust. , London
- 3.Fournier D, Chirouze C, Leroy J. (2013) Alternatives to Carbapenams in ESBL-producing Escherichia coli infections. , Med Mal Infect 43, 62-6.
- 4.Esteve-Palau E, Solande G, Sanchez F. (2015) Clinial and economic impact of urinary tract infections caused by ESBL-producing Escherichia coli requiring hospilization: a matched cohort study. , J Infect 71, 667-74.
- 5.Roschanski N, Fischer J. (2014) Development of a multiplex real-time PCR for the rapid detection of the predominant beta-lactamase genes. CTX-M, SHV, TEM and CIT-Type AmpCs in Enterobacteriaceae. PLoS ONE. 9 100956.
- 6.Shin J, Ko K. (2015) Effect of plasmids harbouring blaCTX-M on the virulence and fitness of Escherichia coli ST131 isolates. , J Antimicrob Agents 46, 214-18.
- 7.Yang Y, Ku C, Lin J.(201) Impact of extended-spectrum β-lactamase-producing Escherichia coli and Klebsiella pneumoniae on the outcome of community-onset bacteremic urinary tract infections. , J Microbiol Immunol Infect 43, 194-9.
- 8.Swayne R, Ellington M, Currna M. (2013) Utility of a novel TaqMan PCR assay for metallo-B-lactamase genes plus other TaqMan assays in detecting genes encoding serine carbapenamases and clinically significant extended-spectrum B-lactamases. Antimicrob Agents.42. 352-6.
- 9.Birkett C, Ludlam H, Woodford N. (2007) Real-Time TaqMan PCR for rapid detection and typing of genes encoding CTX-M extended-spectrum B-Lactamases. , J Med Microbiol 56, 52-5.
- 10.Wintermans B, Reuland E, Wintermans R. (2013) The cost-effectiveness of ESBL detection: towards molecular detection methods?. , Clin Microbiol Infect 19, 662-5.
- 11.Lupo A, Papp-Wallace K, R Sendi Bonomo. (2013) Non-phenotypic tests to detect and characterize antibiotic resistance mechanisms in Enterbacteriaceae. , Microbiol Infect Disl 77, 179-94.
- 12.Zee A Van der, Roorda L, Bosman G. (2016) Molecular diagnosis of urinary tract infections by semi-quantitative detection of uropathogens in a routine clinical hospital setting. , PLoS ONE 11, 0150755.
- 13.Singh P, Pfeifer Y, Mustapha A. (2016) Multiplex real-time PCR assay for the detection of extended-spectrum B-lactamase and carbapenamase genes using melting curve analysis. , J Microbiol Methods 124, 72-8.
- 15.Newcombe R. (1998) Two sided confidence intervals for the single proportion: a comparative evaluation of seven methods. , Statistics in Medicine 17, 857-92.
- 17.Yazdi M, Nazemi A, Mirinargasi M. (2012) Genotypic versus phenotypic methods to detect extended-spectrum beta-lactamases (ESBLs) in uropathogenic Escherichia coli. , Ann Biol Res 3, 2454-8.
Cited by (3)
- 1.Olorunleke Solomon Olabiyi, Kirchner Miranda, Duggett Nicholas, Stevens Kim, Chah Kennedy F., et al, 2024, Rapid detection and molecular epidemiology of β-lactamase producing Enterobacteriaceae isolated from food animals and in-contact humans in Nigeria, PLOS ONE, 19(4), e0289190, 10.1371/journal.pone.0289190
- 2.Sultan Asfia, Khan Fatima, 2022, , , (), 205, 10.1007/978-981-16-9097-6_13
- 3.Bidet Philippe, Birgy André, Ouldali Naim, Béchet Stéphane, Levy Corinne, et al, 2022, Comparative genomic analysis of ESBL-producing Escherichia coli from faecal carriage and febrile urinary tract infection in children: a prospective multicentre study, JAC-Antimicrobial Resistance, 4(3), 10.1093/jacamr/dlac056