
Glass Chromatography Application: TLC Separation of Benzoic Esters in Pharmaceutical Products
- Open Access
- Peer Reviewed
- Similarity Checked
- CC BY 4.0
Abstract
p-hydroxybenzoic acid esters are used as food and drug preservatives. These compounds were quantised by a reversed-phase thin-layer chromatography method based on the use of silanized silica gel as stationary phase. Thin layers chromatography of silanized silica gel (HF254) is implemented to separate p-hydroxybenzoic acid and its methyl, ethyl, propyl, butyl and benzyl esters. Borate buffer (pH 2) was used as a mobile phase with the addition of organic solvent as required. For the quantitative determination, the solutions to be analysed were applied in bands on 5 x 20 cm plates. The plates are developed in glass chromatography chambers lined with filter paper. After the plates have been developed they are dried at room temperature. The spots or bands of the various compounds are visualised under a 250-mµ UV light source. The extraction of the silica gel with methanol was effective. Six preservatives were separated with better results for benzyl- and butyl-p-hydroxybenzoates. Chromatographic development controlled by temperature stability in the chromatographic chamber and spectrophotometric determination of all the compounds were indicated. A second development with the same solvent mixture was suggested especially when low RF is involved. Various compounds are completely separated and a good determination of p-hydroxybenzoic acid and its principle esters are possible using a simple technique of elution and spectrophotometric determination.
Article Information
- Received
- Accepted
- Published
Academic Editor: Solomon Genet, School of Medicine, Addis Ababa University
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2017 Loai Aljerf, et al.
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Corresponding author: Correspondence: Loai Aljerf, Department of Life Sciences, Faculty of Dentistry, University of Damascus, Syria. Phone: +963 944 48 22 03 —
Competing Interests
The authors have declared that no competing interests exist.
Funding
No specific funding statement was provided by the authors.
Data Availability
No data-availability statement was provided by the authors.
Citation:
Introduction
The increasing use of p-hydroxybenzoic acid esters as food and drug preservatives has led to the development of many analytical techniques for identifying and assaying these compounds.1, 2, 3, 4 For several years, thin-layer chromatography (TLC) in particular has often been used with good results.5, 6 In this report we shall briefly describe a reversed-phase TLC method based on the use of silanized silica gel as stationary phase. Coupled with a simple elution and spectrophotometric determination technique, it has proved highly effective in separating and determining p-hydroxybenzoic acid and its principal esters.
Experimental
Preparation of the Chromatographic Plates
About 30 g of HF254 (Merck) silanized silica gel were suspended in 60 mL of a (2 : 1) water-methanol mixture by shaking vigorously for a few minutes in order to obtain a homogeneous suspension. The latter was then distributed on the plates using a Desaga spreader so as to obtain a uniform layer of 0.25 mm thickness. The plates were dried in air for about 3 h and stored over silica gel in a desiccator. They should not be used for at least 24 h after preparation.
Preparation of the Chromatography Solutions
Methanol solutions of pure commercial products (Merck, BDH, Carlo Erba) were used as reference solutions for p-hydroxybenzoic acid and its esters. The method is proper to extract the preservatives from pharmaceutical specialities and to prepare the sample solutions.
Application of the Solutions to the Plates
To identify the substances, the sample and reference solutions were applied as spots in parallel on the same plate using a micropipette and taking care to pipette no more than 5 µL each time, and to deposit from 2 to 10 µg of each preservative on the plate. For the quantitative determination, the solutions to be analysed were applied in bands on 5 x 20 cm plates, pipetting from 0.025 to 0.1 mL of solution so that from 20 to 100 µg of each substance are applied to each plate. The solutions were applied 2 cm from the starting edge leaving a margin of approx. 0.5 cm on both sides of the plate.
Chromatographic Development
Chromatographic development was carried out using borate buffer, pH 2 (502 mL of 0.2 M H3BO3 solution diluted to 1000 mL with 0.2 M NaOH solution) as mobile phase, adding a given volume of organic solvent as required. The following buffer-solvent combinations have proved most suitable for separation of the various preservatives: buffer-methyl alcohol (90 : 10), buffer-ethylcellosolve (90 : 10), buffer-ethyl acetate (saturated solution at 25 OC), buffer-ethyl ether (saturated solution at 15 OC), buffer-tetrahydrofuran (95 : 5), buffer-dioxane (90 : 10).
The plates are developed in glass chromatography chambers lined with filter paper. Before putting the plates in them, the chrormatographic chambers were saturated with the solvent vapours for about 1 h. The plates are allowed to develop until the solvent front has risen about 14 cm from the point where the substance was deposited (90 to 120 min). When double development is desired, the plate is dried at room temperature for about 1 h and then returned to the chromatographic chamber. Chromatographic development is performed at about 25 OC except when the mixture contains ethyl ether in which case the temperature of the chromatographic chamber should be 15 OC.
Detection of the Compounds
After the plates have been developed they are dried at room temperature. The spots or bands of the various compounds were visualised under a 250-mµ UV light source.
Quantitative Analysis
After detection, the band of each substance is isolated by cutting away the sides of the layer of adsorbent with a spatula, and then performing the quantitative determination by removing the silica gel and extracting it with methyl alcohol. A silica gel collection method using a vacuum described by several authors7 has proved useful for this purpose. We have adopted the simple device shown in Figure 1 which permits the easy collection of the silica gel and quick elution of the substances under vacuum. The device consists basically of a small glass funnel inserted, with a special plastic (polyvinylchloride) fitting, into a Pyrex glass volumetric flask (Figure 1A). Near the lower end of the funnel stem is a perforated sintered glass filter plate with gauze or fibre glass pressed onto it; the system can be connected to the vacuum pump by means of the side attachment on the plastic fitting. The silica gel is collected by inserting a small glass tube with a pointed end in the funnel using a tygon ring. The device is turned upside down, and the silica gel is sucked in by applying a low vacuum (Figure 1B). When the silica gel has been collected, a few tenths of mL of methyl alcohol are sucked through to wash the inlet tube (taking care that no solvent emerges from the bottom end of the funnel stem), and the device is quickly uprighted. The vacuum is broken, the inlet tube is removed, 1 or 2 mL of methyl alcohol are pipetted into the funnel, the vacuum is again applied, and the solvent is collected in the volumetric flask. Methyl alcohol is added a couple more times until elution of the substance to be determined is complete (generally after 4-5 mL of methyl alcohol have passed through the funnel, elution is complete).
Figure 1. (1: 2 scale), A = system for elution of substances; B = system for suction of silica gel; a = Pyrex glass funnel with perforated sintered glass filter plate, b = rubber stopper, c = polyvinylchloride fitting with side inlet for vacuum, d = volumetric flask, e = glass tube with tygon ring.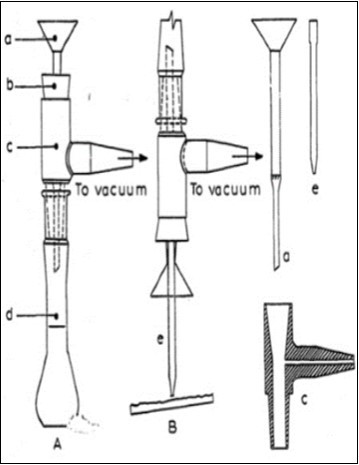
Download figure
The eluent in the volumetric flask is diluted to volume with methyl alcohol and divided into two portions. One was acidified and the other was made alkaline by adding 2% of 1 N HCl and 1 N NaOH, respectively. Light absorption is measured at 297 mµ using the alkaline solution as test solution and the acidified solution as blank following the differential spectrophotometric method.
Results and Discussion
Table 1 shows the RF values obtained on developing the chromatograms with the various buffer-solvent mixtures.
Table 1. Values obtained on HF254 silanized silica gel for the various compounds studied.| Compound | RF valuesa | |||||
| M1 | M2 | M3 | M4 | M5 | M6 | |
| Benzyl-p-hydroxybenzoate | 0.04 | 0.08 | 0.03 | 0.02 | 0.03 | 0.08 |
| Butyl-p-hydroxybenzoate | 0.06 | 0.11 | 0.06 | 0.05 | 0.08 | 0.13 |
| Propyl-p-hydroxybenzoate | 0.15 | 0.22 | 0.14 | 0.15 | 0.14 | 0.20 |
| Ethyl-p-hydroxybenzoate | 0.23 | 0.32 | 0.25 | 0.25 | 0.24 | 0.28 |
| Methyl-p-hydroxybenzoate | 0.30 | 0.38 | 0.34 | 0.35 | 0.33 | 0.35 |
| p-hydroxybenzoic acid | 0.60 | 0.70 | 0.069 | 0.75 | 0.57 | 0.59 |
Of the mobile phases studied, mixtures C, D, E, and F gave the best separation of the various compounds, especially of benzyl- and butyl-p-hydroxybenzoates. However, mixtures C and D are rather difficult to use because the solvents are volatile and the solutions are saturated; good chromatographic development and reproducibility of data are closely controlled by temperature stability in the chromatographic chamber. Mixtures E and F are more practical, and we normally use them for the analysis of preservatives in pharmaceutical preparations.8, 9
In quantitative zone chromatography, the separation may not be good enough for elution and quantitative determination of the components of the mixture when the amounts of some of the compounds to be separated are much larger than others. In this case, the mobility of the substances and the chromatographic separation can be greatly enhanced by performing a second development with the same solvent mixture, especially when low RF paraseptics are involved.
Table 2 shows the RF’ values obtained with mixtures E and F after a second development. In this case the RF’ values represent the proportion between the distance of the spots of substance from the point of application after the second development and the distance of the solvent front from the point of application (the solvent run was the same in the first and second development).
Table 2. RF values calculated after second development on HF254 silanized silica gel.| Compound | RF valuesb | |
| M5 | M6 | |
| Benzyl-p-hydroxybenzoate | 0.06 | 0.17 |
| Butyl-p-hydroxybenzoate | 0.14 | 0.25 |
| Propyl-p-hydroxybenzoate | 0.24 | 0.38 |
| Ethyl-p-hydroxybenzoate | 0.36 | 0.49 |
| Methyl-p-hydroxybenzoate | 0.47 | 0.58 |
| p-hydroxybenzoic acid | 0.68 | 0.75 |
The quantitative analysis of the eluents by differential spectrophotometry eliminates background absorption due to the extraction of the silica gel with methanol. The amount of the test compounds may be calculated from the spectrophotometric results using a calibration straight line plotted from results of chromatographic analyses performed on solutions containing known amounts of preservatives.
Greater precision in quantitative analyses may be obtained by using the internal standard method. In this case a known amount of a compound with a different RF from those of the test compounds is added to the test solution before chromatography (since the six preservatives discussed here are not generally used together in the same pharmaceutical speciality, one which is known to be absent in the test sample may be used as an internal standard). After chromatographic development and spectrophotometric determination of all the compounds, it is easy to calculate the amount of each single compound in the test solution using the formula:
 mg of compound contained in the sample solution
mg of compound contained in the sample solution
Where, P is the amount of internal standard added, expressed in mg; EC and Est are the differential absorption of the eluents of the test compound and of the compound used as internal standard respectively; F is a correction factor calculated from the ratio between the differential absorptions of equal amounts of the internal standard and test compounds (experimentally, the F factor is determined by repeated quantitative chromatographic analyses of mixtures containing known amounts of the two compounds).
Table(a) and (b) show the results of a quantitative chromatographic determination performed on solutions with different concentrations of methyl-p-hydroxybenzoate and propyl-p-hydroxybenzoate using the calibration straight line method and the internal standard method.
The data given in Table 3 show that the quantitative analysis, especially using the internal standard method, is quite accurate; the internal standard method is also more precise than the calibration straight line method because the quantitative determination is not affected by pipetting errors which may occur when the solutions are applied to the chromatographic plates.
Table 3. Quantitative chromatographic analysis of (mean±SD) methanol solutions containing different concentrations of methyl and propyl-p-hydroxybenzoate.| Solution No. | Theoretical (mg/mL) | Foundc | |||
| Method A | Method B | ||||
| mg/mL | % Difference | mg/mL | % Difference | ||
| (a) Methyl-p-hydroxybenzoate | |||||
| 1 | 0.424 | 0.426±0.053 | +0.5 | 0.424±0.038 | 0.0 |
| 2 | 0.824 | 0.807±0.097 | -2.1 | 0.836±0.065 | +1.5 |
| 3 | 1.227 | 1.196±0.101 | -2.5 | 1.260±0.113 | +2.7 |
| 4 | 1.595 | 1.552±0.205 | -2.7 | 1.585±0.190 | -0.6 |
| 5 | 2.006 | 1.992±0.273 | -0.7 | 2.046±0.284 | +2.0 |
| (b) Propyl-p-hydroxybenzoate | |||||
| 1 | 2.008 | 2.045±0.289 | +1.8 | 2.053±0.309 | +2.2 |
| 2 | 1.621 | 1.580±0.173 | -2.5 | 1.613±0.191 | -0.5 |
| 3 | 1.206 | 1.164±0.132 | -3.5 | 1.211±0.138 | +0.4 |
| 4 | 0.804 | 0.781±0.101 | -2.9 | 0.807±0.076 | +0.4 |
| 5 | 0.419 | 0.420±0.036 | +0.2 | 0.416±0.037 | -0.7 |
| Current study | Bajaj and John11 | |
| Ion-exchanger | HF254 silanized silica gel | HF254 silanized silica gel |
| Mobile phase | Borate buffer | Hexane: 1, 4 dioxane: methanol: triethylamine (7.5:1:1:0.5, v/v) |
| Scanning densitometry at λmax | 250±1 nm | 254 |
| Development distance | 12 cm | 1.5 cm |
| Development time | 25 min | 30 min |
| Concentration range | 8-150 μg/ml | Nd |
| Equation | OD = -1.093 + 1.127 C | Nd |
| r | 0.9996 | Nd |
| Mean accuracy | 99.16±0.52% | Nd |
| LOD | 0.24 µg per zone | 0.80 µg per zone |
| LOQ | 0.43 µg per zone | Nd |
A repeatability test consisting of 15 chromatographic determinations on a methyl- and propyl-p-hydroxybenzoate mixture showed that the coefficient of variation for both paraseptics is about 3% for the calibrated straight line method and about 1.5% for the internal standard method.
The current method relies on the use of inexpensive equipment, a scanner and software, and does not use of critical derivatizing reagent, thus maximizing the ability of laboratories worldwide to analyze pharmaceutical samples. It also specifies a rapid separation of Benzyl-p-hydroxybenzoate, Butyl-p-hydroxybenzoate, Propyl-p-hydroxybenzoate, Ethyl-p-hydroxybenzoate, Methyl-p-hydroxybenzoate, p-hydroxybenzoic acid. Moreover, the method is accurate and sensitive stability-indicating TLC method based on separation of p-hydroxybenzoate from its samples at ambient temperature 25±5°C and detection at 250nm in an overall analysis time of about 3 min.
Conclusion
Hydroxybenzoates are completely separated including a proper determination of p-hydroxybenzoic acid. The concentrations of p-hydroxybenzoic acid and its principle esters in food and pharmaceutical products can possibly being chromatographically measured using a simple technique of TLC elution and spectrophotometric determination.
References
- 1.Gurina D L, Antipova M L, Odintsova E G, Petrenko V E. (2017) Solvation of para-hydroxybenzoic acid and its esters (methylparaben, propylparaben) in supercritical carbon dioxide. Computer simulation. , J Supercrit Fluids 120, 59-64.
- 2.Goto A, Miyamoto H, Odintsova E G, Petrenko V E. (2013) ChemInform Abstract: IUPAC-NIST Solubility Data Series. Part 90. Hydroxybenzoic Acid Derivatives in Binary and Ternary Systems. Part 2. Hydroxybenzoic Acids, Hydroxybenzoates, and Hydroxybenzoic Acid Salts in Nonaqueous Systems.DOI:. 10-1002.
- 3.Tran T M, Minh T B, Kumosani T A, Kannan K. (2016) Occurrence of phthalate diesters (phthalates), p -hydroxybenzoic acid esters (parabens), bisphenol A diglycidyl ether (BADGE) and their derivatives in indoor dust from Vietnam: Implications for exposure. , Chemosphere 144, 1553-1559.
- 4.Beltyukova S V, Malinka E V, Liventsova E O. (2016) Determination of para-hydroxybenzoic acid and its esters in solid phase of sorbent with using of sensitized luminescence of Tb(II). in complex with 2,2’-dipyridyl. Chemistry 21(4), 21-30.
- 5.Volkmann D. (1980) Separation of p-hydroxybenzoic acid esters by circular high performance thin-layer chromatography. , J. High Resolut. Chromatogr 3(4), 189-190.
- 6.Shtykov S N, Sumina E G, Uglanova V Z, Berezkin V G. (2016) Thin-layer chromatography of benzoic acids with a controlled gas phase: A comparison of different stationary phases. , J. Planar Chromat 29(1), 66-71.
- 7.Aljerf L, Choukaife A E. (2017) A novel method to chromatographically resolution of sulphonamides by vapour - programmed Thin - Layer Chromatography. , MOJ Bioorganic Org. Chem 1(4), 00024-10.
- 8.Raof S, Mohamad S, Mhd Abas. (2013) Synthesis and Evaluation of Molecularly Imprinted Silica Gel for 2-Hydroxybenzoic Acid in Aqueous Solution. , Int. J. Mol. Sci 14(3), 5952-5965.
- 9.Mirzaie A, Jamshidi A, Husain S. (2007) Quantitative ion-exchange TLC ofp-hydroxybenzoic acid in the presence of preservatives. , J. Planar Chromat 20(4), 303-306.