
A New Gene Mutation of PRKAR1A was found in a Carney Complex Case
Abstract
A patient with Carney complex is reported with a previously undescribed PRKAR1A mutation. The article situates the variant within PKA pathway biology and clinical phenotype, underscoring the value of genetic testing for diagnosis, counseling, and surveillance.
Article Information
- Received
- Accepted
- Published
Academic Editor: Aroma Oberoi, Professor & Head, Department of Microbiology, Christian Medical College & Hospital
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2021 Hai-Xuan Ding, et al.
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Corresponding author: Li Yang, Department of Endocrinology, Hunan Provincial People's Hospital (The First Affiliated Hospital of Hunan Normal University), No. 61 Jiefangxi Road, Furong District, Changsha 410000, Hunan Province, China. Tel: +86 15073167059 —
Competing Interests
The authors have declared that no competing interests exist.
Funding
This work was supported by the Foundation of Hunan Provincial Health and Family Planning Commission (No. B2019067), the Natural Science Foundation of Hunan Province (No. 2021JJ30404), and the 2019 Renshu Foundation Project (No. RS201910).
Data Availability
No data-availability statement was provided by the authors.
Citation:
Introduction
Carney complex (CNC) is a rare multiple tumor syndrome characterized by patchy pigmentation of the skin and mucous membranes associated with a variety of non-endocrine and endocrine tumors, including Primary Pigmented Nodular Adrenocortical Disease (PPNAD). Carney complex is an autosomal dominant multiple tumor syndrome 1. It has been reported that most PPNAD or Carney syndrome is associated with mutations in the PRKAR1A gene, in addition to mutations in the PDE11A and PDE8B genes 2, 3. We report a case of PPNAD-associated Cushing's syndrome diagnosed as CNC due to a novel genetic causative locus in PRKAR1A. This is a novel mutation that has not been publicly reported and it is associated with PPNAD.
Case Report
A 13-year-old boy presented 2 years ago with facial rounding and hyperpigmentation of the face, eyelids and lips. Growth was slow, approximately 3 cm/2 yr. Six months ago, the patient developed acne on the face. Half a month ago, the patient felt pain in the lower back and was affected by activity. Past history: postural lithotripsy was performed in August 2017 and April 2018. Family history: The family complained of hyperpigmentation on the face and lips of the father, aunt and grandmother, who did not have a similar obese body type, but they could live a normal life. Physical examination: height 148 cm, weight 70 kg (BMI 31.95 kg/m2, BP 120/80 mmHg, full moon face, obese body type, upper body measurement 66 cm, lower body measurement 82 cm, finger spacing 142 cm, patchy pigmentation visible on mouth, lips and eyelids, facial acne (Figure 1 BCD), acanthosis nigricans visible on neck and axillae, breast Tanner stage 3 (Figure 1 E). Pubic hair growth, short penis, normal testicular development, Tanner stage 3, red rash was seen in the groin, and the toes became rough and grayish white. No obvious abnormalities were found in the heart, lungs and abdomen.
Laboratory findings showed hypercortisolism, and cortisol levels remained high after a 1 mg dexamethasone suppression test (Table 1), suggesting that Cushing's syndrome is not related to ACTH.
Table 1. Patient laboratory parametersAdrenal computed tomography (CT) showed multiple small nodules seen in the left adrenal inner branch and body (Figure 2 A), which were considered to be adenomas or adrenal tuberculosis; enhanced CT of the pituitary gland did not show any abnormalities. Magnetic resonance imaging of the thoracic spine showed a compression fracture of the thoracic spine (T11) and whole-body bone SPECT showed a compression fracture of the 11th thoracic vertebra; echocardiography did not show any cardiac mucosal tumor. Combined with the patient's history and physical examination, small adrenal nodular hyperplasia not dependent on ACTH hyperplasia is currently considered.
The patient underwent laparoscopic resection of the left adrenal gland in the Department of Urology and was sent for pathological examination (Figure 2 B) of the left adrenal gland, a grayish-yellow tissue measuring 8*6*3.5 cm (Figure 2 D). Genetic analysis of the patient revealed the mutated gene PPKAR1A variant locus C.1-2942 G>A (Figure 3). Through a series of tests, the patient was diagnosed with Carney syndrome and a new mutated locus for the causative gene was identified. The patient's ACTH was 0.488 ↓ pg/ml (7-64) and plasma cortisol was 14.38 ug/dl (4.26-24.85) at the 8 a.m. postoperative follow-up on the fourth day. The patient was treated with prednisone 20 mg Qd after discharge from the hospital. At 10 months postoperatively, the patient was followed up and grew 5 cm taller and lost 5 Kg in weight compared to the time of admission . At 18 months postoperatively, the patient's ACTH and plasma cortisol had normalized at the time of review, and he had reached a height of 158 cm and a weight loss of 68 kg .
Figure 1. (ABCDEF) Physical signs of the patient before adrenalectomy. Cushing-like features include moon face before surgery (BC), central obesity (F), breast development (E), spots on the face, lips, oral mucosa and skin pigmentation (CD). 
Download figure
Figure 2. Computed tomography of the patient's adrenal gland showed multiple small nodules in the left adrenal inner branch and body (Figure A), considering adenoma or adrenal tuberculosis. The patient's left adrenal gland macroscopically showed a piece of grayish-yellow tissue measuring 8*6*3.5 cm, including an adrenal gland measuring 6.5*2.5*0.3 cm, with multiple small nodules of 0.2-0.7 cm in diameter attached to the surface (C). Microscopic examination of the patient's left adrenal gland showed multiple nodules in the adrenal cortex, without envelope, partially protruding from the adrenal gland, with clear cytoplasm and eosinophilic cells (D). 
Download figure
Figure 3. DNA sequence detection: variant gene PPKAR1A variant locus C.1-2942G>A, associated disease: Cushing's syndrome (primary pigmented nodular adrenocortical disease type 1, PPNAD1). 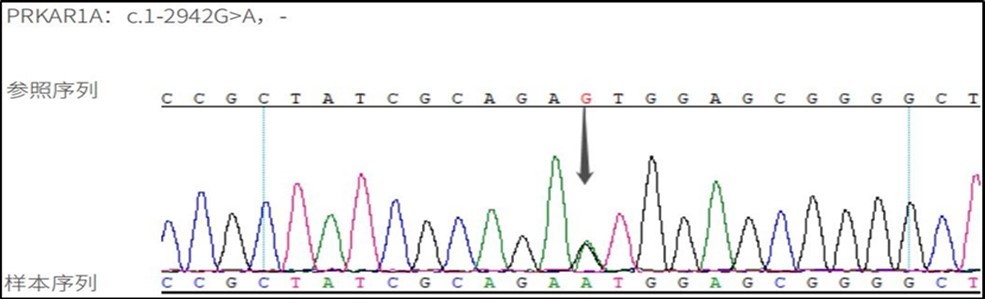
Download figure
Discussion
In the above case report, we describe a patient with typical features such as patchy pigmentation of the face, lips and oral mucosa, compression fractures of the thoracic spine leading to osteoporosis and PPNAD, and the discovery of the PPKAR1A variant gene by genetic testing, meeting the diagnostic criteria for CNC (Figure 3).
PPNAD is a rare non-adrenocorticotropin-dependent Cushing's syndrome 4. It has a characteristic histological presentation of multiple small (<1 cm) dark brown nodules scattered throughout the cortex, and the diagnosis is difficult to make without histological examination, as PPNAD has mostly normal or even small adrenal glands on adrenal imaging 5, and this patient's left adrenal gland showed multiple small nodules on CT in the inner branches and corpora. Treatment recommendations for PPNAD vary, with many authors recommending bilateral adrenalectomy 6, 7, 8, while others believe that in some cases, unilateral adrenalectomy can achieve clinical and biochemical improvement, and that removal of a second adrenal gland should be considered when cortisolism recurs 6, 9. Meanwhile, the Endocrine Society's 2015 clinical practice guidelines recommend one-stage surgical resection for bilateral adrenal disease 10, and our patient opted for left adrenalectomy due to being in the growth phase, and was given a postoperative hormone replacement regimen with continued follow-up at a later stage and contralateral adrenalectomy if necessary.
PPNAD is one of the manifestations of Carney complex (CNC), which was first reported by JA Carney in 1985 1. Already in 2000, it was shown that inactivating germline mutations in the PRKAR1A gene encoding the protein kinase A (PKA) 1a regulatory subunit (RIa) were found in most CNC and PPNAD patients, resulting in increased PKA activity due to PRKAR1A mutations in CNC11, 12. CNC is a rare multiple tumor with additional clinical features of skin pigmentation, cardiac mucinous tumor, neurological and endocrine tumors can be diagnosed from the patient's clinical presentation, laboratory findings and/or genetic examination in a 13-year-old boy patient with histological confirmation of PPNAD in resected left adrenalectomy and inactivated PRKAR1A alterations in genetic sequencing Carney complex. Pathogenic variants in the PRKAR1A gene are present in more than 70% of CNC patients and up to 80% of PPNAD patients with combined Cushing's syndrome 13, the PRKAR1A gene is located on chromosome 17q24.2, and there are at least 140 known types of PKAR1A pathogenic mutations (http://prkar1a.nichd.nih.gov/hmdb/intro.html), most of the germline mutations were located in exons, especially exons 2, 3, 5, 7 and 8, and a few (20%) were located in intron sequences, affecting splicing 14. This patient was detected with a variant in the PRKAR1A gene at locus c.1-2924G > A. A 1-locus variant occurred, consistent with primary pigmented nodular adrenocortical disease type 1, which is autosomal dominant, and a heterozygous locus variant was detected in the PRKAR1A gene associated with it, which is rare and has been poorly studied, so pathogenicity is unclear, but The possibility of causing the disease cannot be excluded. A search of public databases such as ClinVar showed that the c.1-2924G>A variant is located in the UTR region, and this variant is not included in the Chinese population of the Thousands Project, the East Asian population of the EXAC database, the East Asian population of the gnomAD database, or local databases. Groussin L et al 15 reported this locus and 11 patients with carney complex participated in the study, all of whom had symptoms of primary pigmented nodular adrenocortical disease of Cushing's syndrome. They were genetically tested and the locus was detected in 1 patient, and this paper indicates that the variant forms a new start codon. Western blotting was performed using the mouse primary antibody RIa subunit, and PCR cloning methods were used to construct wild-type and exon 1B mutant expression structures. sequencing of PRKAR1A exon 1B revealed a heterozygous mutation that created an upstream, out-of-frame RIa1B mRNA within the consensus sequence of the ATG codon for translation initiation, and according to a scanning model of eukaryotic translation, the new ATG should initiate translation of a truncated protein and reduce translation of the codon from the wild type start. The PRKAR1A defects reported so far are all functional defects a pair of null mutations, and all sequence changes are predicted to result in a premature termination codon except for one mutation that alters the transcription initiation site (ATG codon). It was subsequently demonstrated that mutant mRNAs carrying premature termination codons are unstable as a result of nonsense-mediated mRNA decay (NMD), but the most important finding in the study was that altered PRKAR1A function (not only its complete deletion) is sufficient to enhance PKA activity, leading to tumorigenesis in CNC-affected tissues 15. Gene sequencing revealed a new pathogenic variant in the PRKAR1A gene due to sequencing, our patient was found to have a new shifted code pathogenic variant (c.1-2942G>A) leading to the formation of a new start codon, he had PPNAD and some scattered hyperpigmentation, but the current clinical history and examination did not match other features of CNC, and even then should be considered in all PPNAD CNC should be considered in all patients with PPNAD, and timely diagnosis and regular monitoring of CNC manifestations may help to prevent complications of the disease, In particular, complications from cardiac tumors, cardiac mucinous neoplasms affect 20-40% of CNC resulting in embolic stroke, heart failure and arrhythmias. Recommendations for CNC screening tests include echocardiography (annually or every two years depending on the history of cardiac mucinous neoplasms), skin assessment, thyroid ultrasound, pituitary MRI, testicular/ovarian ultrasound, and growth hormone, insulin-like growth factor 1 and prolactin serum measurements 16.
Conclusion
Our cases increase the number of reported cases of CNC, a disease with the potential for late development of cardiac mucinous tumors, and these patients must be evaluated clinically followed by long-term follow-up. Also our current cases of Cushing's syndrome, in which the diagnosis of CNC is established due to a novel predicted inactivating pathogen in the PRKAR1A gene, have led to an emphasis on this gene mutation and improved eugenics. The early diagnosis of endogenous cortisolism remains a diagnostic challenge, increasing awareness of the disease and improving clinicians' knowledge of the disease, facilitating early identification and timely intervention of the disease. The novel mutations presented in this paper are considered to be causative factors of PPNAD, and timely diagnosis of CNC and careful surveillance can help prevent potentially fatal complications of the disease.
Compliance with ethical standards
Ethical Approval
This article does not contain any studies with animals and human participants performed by any of the authors.
Informed Consent
Informed consent was obtained from all individual participants included in the study. The patient and his parents provided informed consent for the release of this case report and images.
References
- 1.J A, Carney. (1985) The complex of myxomas, spotty pigmentation, and endocrine overactivity. , Medicine (Abingdon) 13(1), 19-26.
- 2.Anelia Horvath, Sosipatros Boikos, Christoforos Giatzakis. (2006) A genome-wide scan identifies mutations in the gene encoding phosphodiesterase 11A4 (PDE11A) in individuals with adrenocortical hyperplasia. , J .Nat Genet 38, 794-800.
- 3.Anelia Horvath, Christoforos Giatzakis, Kitman Tsang. (2008) A cAMP-specific phosphodiesterase (PDE8B) that is mutated in adrenal hyperplasia is expressed widely in human and mouse tissues: a novel PDE8B isoform in human adrenal cortex. , J .Eur J Hum Genet 16, 1245-1253.
- 4.A De Venanzi, Alencar G A, Bourdeau I. (2014) Primary bilateral macronodular adrenal hyperplasia. Current Opinion in Endocrinology Diabetes & Obesity 21(3), 177-184.
- 5.Rockall A G, Babar S A, SAA Sohaib. (2004) CT and MR imaging of the adrenal glands in ACTH-independent cushing syndrome. Radiographics A Review Publication of the Radiological Society of North America Inc. 24(2), 435-452.
- 6.Groussin L, Cazabat L, René. (2005) Adrenal pathophysiology: lessons from the Carney complex. Horm Res Paediatr. 64(3), 132-139.
- 7.Sikorska D, Bednarek-Papierska L, Mojs E. (2017) Samborski W Bilateral primary pigmented nodular adrenal disease as a component of Carney syndrome - case report. Endokrynol Pol. 68(1), 70-72.
- 8.Lowe K M, Young W F, Lyssikatos C. (2016) Cushing Syndrome in Carney Complex: Clinical, Pathologic, and Molecular Genetic Findings in the 17 Affected Mayo Clinic Patients. Am J Surg Pathol. 41(2), 171-181.
- 9.Wei Q, Jin X L, Zhu Y B. (2006) Primary pigmented nodular adrenocortical disease: a report of 5 cases. , Journal of Diagnostics Concepts and Practice 119(9), 782-785.
- 10.Nieman L K, BMK Biller, Findling J W. (2015) Treatment of Cushing's Syndrome: An Endocrine Society Clinical Practice Guideline. , J Clin Endocrinol Metab 8, 2807-2831.
- 11.L S Kirschner, J A Carney, S D Pack. (2000) Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. , J Nat Genet 26, 89-92.
- 12.L S Kirschner, Sandrini F, Monbo J.Genetic heterogeneity and spectrum of mutations of the PRKAR1A gene in patients with the carney complex. , J Hum Mol Genet 2000, 3037-46.
- 13.Jérôme Bertherat. (2009) Horvath Anélia, Groussin Lionel et al. Mutations in regulatory subunit type 1A of cyclic adenosine 5'-monophosphate-dependent protein kinase (PRKAR1A): phenotype analysis in 353 patients and 80 different genotypes. , J Clin Endocrinol Metab 94, 2085-2091.
- 14.Amit Tirosh. (2018) Valdés Nuria, Stratakis Constantine A, Genetics of micronodular adrenal hyperplasia and Carney complex. , J Presse Med 47-127.
- 15.Groussin L, L S Kirschner, Vincent-Dejean C. (2002) Molecular Analysis of the Cyclic AMP-Dependent Protein Kinase A (PKA). Regulatory Subunit 1A ( PRKAR1A ) Gene in Patients with Carney Complex and Primary Pigmented Nodular Adrenocortical Disease (PPNAD) Reveals Novel Mutations and Clues For Pathophysiology: American Journal of Human Genetics 71(6), 1433-1442.