
The Innermore Ossein- Enchondroma
Abstract
Preface Frequently, benign bone tumours are an incidental discovery wherein clinical symptoms are contingent to tumour location and magnitude and manifest as localized pain, swelling, skeletal deformities or pathologic fracture. Radiographic imaging is a cogent methodology of discerning the neoplasms.
Article Information
- Received
- Accepted
- Published
Academic Editor: Qiping Dong, China.
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2021 Anubha Bajaj
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Corresponding author: Anubha Bajaj, Consultant Histopathologist at A.B, Diagnostics at A-1, Ring Road, Rajouri Garden, New Delhi 110027 —
Competing Interests
The authors have declared that no competing interests exist.
Funding
No specific funding statement was provided by the authors.
Data Availability
No data-availability statement was provided by the authors.
Citation:
Preface
Benign bone tumours are a frequent, incidental discovery wherein clinical symptoms are contingent to tumour location and tumour magnitude. The neoplasms manifest on-site pain, localized swelling, skeletal deformities or pathological fractures. Radiographic imaging is a cogent methodology for discerning the neoplasms. Enchondroma is a benign, lobulated neoplasm engendered from hyaline cartilage and is commonly discerned within medullary cavity of bones of endochondral origin. The neoplasm lacks a definitive pattern of genetic inheritance.
Disease Characteristics
Enchondroma is an intraosseous, cartilaginous neoplasm with an estimated prevalence of 3% of comprehensive bone tumours and around 13% of benign bone neoplasms. Enchondroma is usually represented as a solitary (70%), centric, metaphyseal neoplasm predominantly occurring within miniature, tubular bones of hands or feet or long bones such as femur or humerus although no site of tumour emergence is exempt. The neoplasm is exceptionally denominated within thumb or ribs 1, 2.
Classically, enchondroma arises during the second decade between 15 years to 35 years although no age of disease emergence is exempt. Tumefaction may also appear within childhood. The neoplasm depicts an equivalent gender prevalence1, 2.
Enchondroma is frequently discerned within miniature tubular bones of the hand. Classically, enchondroma is situated within proximal metaphysis of proximal phalanx with a predilection for ulnar surface of the hand 1, 2.
Expansible, medullary cartilaginous neoplasms may terminate in pathological fractures due to encroachment upon the cortical bone. Solitary enchondoma is accompanied by malignant transformation in around <1% instances. Malignant metamorphosis into a chondrosarcoma usually occurs following skeletal maturity 1, 2.
Disease Pathogenesis
Tumefaction emerges from ectopic rests of growth plate cartilaginous cells or chondrocytes which proliferate, enlarge and persist during adulthood although further evolution ceases. A displaced growth plate may generate an enchondroma1, 2.
Enchondroma is associated with exceptional, sporadic, somatic mutations within isocitrate dehydrogenase-1 (IDH1) and isocitrate dehydrogenase- 2 (IDH2) genes. Isocitrate dehydrogenase is an enzymatic component of tricarboxylic acid (TCA) cycle which converts isocitrate to alpha-ketoglutarate1, 2.
Mutations within IDH1 and IDH2 genes engender an enzymatic malfunction with consequently enhanced levels of onco-metabolite D-2-hydroxyglutarate (D-2-HG.). The molecule D-2-HG competitively inhibits alpha-ketoglutarate dependent enzymes1, 2.
Subsequent hyper-methylation of deoxyribonucleic acid (DNA) and modification of histones ensue which may influence cellular differentiation. Inhibited osteogenic differentiation of mesenchymal stem cells occurs on account of elevated D-2-HG levels arising due to genetic mutations within IDH1 and IDH2 genes. In essence, prohibition of osteogenic differentiation during skeletal configuration engenders the cartilaginous neoplasm 1, 3.
Clinical Elucidation
Enchondroma is frequently associated with nonspecific clinical symptoms. Tumour discernment usually occurs due to a pathological fracture secondary to bone trauma with accompanying localized or generalized, radiating pain. Several neoplasms are incidentally discovered upon cogent radiographic imaging 3, 4.
Majority of enchondromas commence within medullary fraction of bony diaphysis. The neoplasm generated within metaphyseal region demonstrate an extraneous cortical expansion. Enlarged lesions may engender a pathological fracture 3, 4.
Histological Elucidation
On gross examination, enchondroma appears as a grey/blue, translucent, hypo-cellular, non-vascular neoplasm imbued with abundant hyaline cartilage. Besides, tumefaction may appear as a solid, well circumscribed, cartilaginous, pale blue tumefaction. Foci of myxoid change may be absent 3, 4.
Characteristically, enchondroma is below < 5 centimetre magnitude although enlarged lesions can be considered benign when accompanying indeterminate or atypical microscopic features are absent4, 5. Enchondroma is composed of well-circumscribed nodules of benign hyaline cartilage. Incorporated chondrocytes demonstrate miniature, uniform, spherical nuclei with condensed chromatin. Bi-nucleate cells are infrequent4, 5.
Typically, chondroid matrix of an enchondroma depicts foci of punctate calcification. Tumour cell nuclei are fairly uniform. Mitotic figures are few. Extensively calcified enchondromas are associated with foci of endochondral ossification 4, 5.
The neoplasm is configured of lobules of hyaline cartilage enveloped by perichondrium-encased bone. Tumefaction can be hyper-cellular with bi-nucleate tumour cells. Myxoid change, foci of calcification and endochondral ossification are discerned. Necrosis may ensue on account of avascular cartilaginous component. Tongues of bone appear to extend into subjacent cartilage demonstrating a sharply defined interface confined to the growth plate4, 5.
Benign, juxta-cortical enchondroma arising within miniature, tubular bones as the hand may be hyper-cellular and demonstrate cellular and nuclear atypia 5.
Distinction between benign cartilaginous lesions and atypical cartilaginous tumour or grade 1chondrosarcoma (ACT/CS1) can be challenging, Typically, benign cartilaginous lesions are hypo-cellular, in contrast to hyper-cellular atypical cartilaginous tumour or grade 1 chondrosarcoma 5, 6. Figure 1, Figure 2, Figure 3, Figure 4, Figure 5, Figure 6, Figure 7, Figure 8.
Figure 1. Enchondroma depicting skeletal deformity with tumour nodules confined to small tubular bones of the hand 10.
Download figure
Figure 2. Enchondroma delineating a glistening, grey/white cut surface with an encapsulated, well circumscribed tumour 11.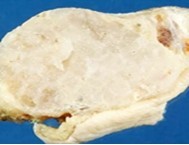
Download figure
Figure 3. Enchondroma exhibiting a nodular, encapsulated, well circumscribed, lobulated appearance 12.
Download figure
Figure 4. Enchondroma depicting lobules of hyaline cartilage with vacuolated cells and eccentric nuclei 12.
Download figure
Figure 5. Enchondroma enunciating lobules of uniform cartilaginous cells with regular, centric nuclei and absence of mitosis 13.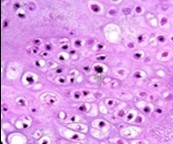
Download figure
Figure 6. Enchondroma exemplifying lobules of hyaline cartilage composed of enlarged, vacuolated cells with uniform nuclei. Focal calcification is absent 14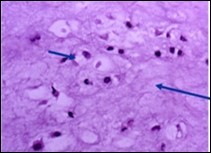
Download figure
Figure 7. Enchondroma enunciating lobular hyaline cartilage with constituent vacuolated chondrocytes an uniform nuclei 15.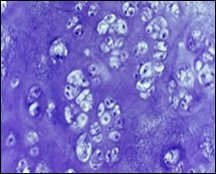
Download figure
Figure 8. Enchondroma delineating hyaline cartilaginous cells with vacuolated cytoplasm and uniform nuclei 16.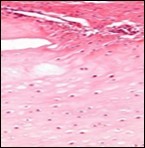
Download figure
Differential Diagnosis
Enchondroma requires a distinction from conditions such as
Bone infarct which demonstrates a radiographic appearance akin to enchondroma. Preliminary lesions depict ghost marrow cells with pyknotic, basophilic nuclei, nucleated red cells, adipose tissue necrosis, focal calcification and necrotic bony trabeculae. Delayed stage exhibits peripheral granulation tissue, gradual layering of new bone upon necrotic bony trabeculae and collagen rimming with focal calcification 5, 6.
Tuberculous dactylitis is associated with pain at initial presentation. Epithelioid cell granulomas are intermingled with focal areas of caseation necrosis and giant cell reaction. Langerhans giant cells may be discerned 5, 6.
Epiphyseal dysplasia usually arises in neonates and children. Multiple joints are simultaneously incriminated. Epiphyseal cartilage is composed of multiple discreet units of chondrocytes segregated by clefts imbued with fibroblast like cells. The cartilaginous component is enveloped within a territorial capsule. Continuity of epiphyseal cartilaginous matrix is disrupted with generation of chondrons or mosaic of chondrocytes 5, 6.
Low grade chondrosarcoma is a malignant neoplasm manifesting pain at initial presentation. Tumefaction erodes superficial bony cortex and depicts a significant myxoid metamorphosis. Enlarged tumours infiltrate bone marrow spaces and appear enmeshed with bony trabeculae. Tumour cells generate a cartilaginous matrix. Atypia may be focal. Intracytoplasmic hyaline globules are commonly discerned within the low grade variant. Columns of cuboidal cells are intermingled within a myxoid stroma 5, 6.
Investigative Assay
On plain radiography, enchondroma depicts a varied appearance which is contingent to location and extent of tumour calcific deposits. The neoplasm may simulate a medullary bone infarct.
Typically, enchondroma appears as a well-defined, solitary defect confined to the metaphyseal region. Centrally situated lesions display well-circumscribed areas of bone rarefaction with a circumscribing, expanded bone cortex, frequently within the diaphysis. Bone cortex is preserved although thinned out. Pathologic fractures may be discerned 7, 8.
Juxta-cortical lesions appear as eccentric, well- demarcated, sub-periosteal, cortical defects. Miniature, flocculent foci of calcification are discerned. Upon radiography, tumour calcification appears as fine, punctate stippling although tumour calcification varies from punctate deposits to calcific rings. Enlarged tumours may engender endosteal scalloping associated with cortical expansion and thinning 7, 8.
Centric neoplasms occur as well-circumscribed lesions with rarefaction of bone diaphysis and expanded bone cortex. Tumours with extensive calcification may simulate a bone infarct, islands of mature bone or a lytic lesion7, 8.
Computerized tomography (CT) can be suitably adopted for discerning mineralization of bone matrix and continuity of bone cortex.
Magnetic resonance imaging (MRI) can detect features indicative of aggressive and destructive neoplasms. Potentially malignant neoplasms are enlarged with significant proportions of un-mineralized component, thinning of adjacent bone cortex and enhanced activity upon bone scan7, 8. Additionally, features such as progressive destruction of chondroid matrix due to an expansive, non-mineralized component, gradually evolving lesion with accompanying pain or an expansible soft tissue tumefaction indicate malignant metamorphosis within an enchondroma.
Magnetic resonance imaging (MRI) of an enchondroma depicts a reduced signal intensity upon T1 weighted imaging along with lobular tumour configuration. Reciprocal, high-intensity modifications appear upon T2 weighted imaging. Peritumoural oedema is absent 8, 9.
Peripheral and spatial image enhancement is observed upon gadolinium contrast imaging. Enchondroma can be suitably differentiated from a bone infarct upon computerized tomography and magnetic resonance imaging 8, 9.
Periostin is a stromal-related protein and is absent in enchondroma. In contrast, alpha-methylacyl-CoA racemase (AMCR) is a mitochondrial and peroxisomal enzyme which is discerned in a majority of enchondromas8, 9.
Therapeutic Options
Conservative management is optimal for benign, asymptomatic bone neoplasms. Curettage and bone grafting is recommended for treating symptomatic, aggressive, non-malignant bone neoplasms8, 9.
Surgical resection may be suitably adopted although employment of either a preliminary or delayed surgical intervention is accompanied by similar treatment outcomes. Inadequate surgical resection may be followed by localized tumour reoccurrence8, 9.
Non surgical treatment of bone lesions can decimate bone structure and integrity. Possibility of occurrence of pathological fracture enhances with magnitude of lesion. Tumefaction exceeding > 2.5 centimetres magnitude, appearing within weight-bearing bones or incriminating an excess of >50% of cortical diameter demonstrate an enhanced probability of fracture. Delayed surgical intervention following fracture union is associated with extended immobilization8, 9. Solitary enchondroma is essentially a self-limited lesion. Curettage and bone grafting is exceptionally followed by tumour reoccurrence. However, enchondroma confined to long bones is associated with enhanced tumour reappearance. Benign enchondroma of long bones may demonstrate localized tumour reoccurrence following surgical extermination8, 9.
References
- 3.Mulligan M E. (2019) How to Diagnose Enchondroma, Bone Infarct. , and Chondrosarcoma” Curr Probl Diagn Radiol 48(3), 262-273.
- 4.Pansuriya T C, R van Eijk, d'Adamo P. (2011) . Somatic mosaic IDH1 and IDH2 mutations are associated with enchondroma and spindle cell hemangioma in Ollier disease and Maffucci syndrome” Nat Genet 43(12), 1256-61.
- 5.Bierry G. (2012) Kerr DA et al” Enchondromas in children: imaging appearance with pathological correlation” Skeletal Radiol. 41(10), 1223-9.
- 6.Zhou X, Zhao B. (2017) The management and surgical intervention timing of enchondromas: A 10-year experience”. , Medicine (Baltimore) 96(16), 6678.
- 7.Amary M F, Damato S. (2011) Ollier disease and Maffucci syndrome are caused by somatic mosaic. mutations of IDH1 and IDH2” Nat Genet 43(12), 1262-5.