
The Minimalistic Malignancy- Low grade Fibromyxoid Sarcoma
Abstract
Low grade fibromyxoid sarcoma (LGFMS) is an exceptional, low grade, soft tissue sarcoma with indolent biological behaviour, extensive preclinical stage, enhanced localized tumour reoccurrence and delayed, distant metastasis. As the deceptively benign neoplasm was initially scripted by Evans in 1987, the tumefaction is nomenclated as “Evan’s tumour”. Incidence of sarcomas is nearly 1% of adult malignancies wherein low grade fibromyxoid sarcoma represents roughly beneath <5% of soft tissue sarcomas 1.
Article Information
- Received
- Accepted
- Published
Academic Editor: Pietro Scicchitano, Cardiology Department, Hospital of Ostuni (BR) - Italy.
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2020 Anubha Bajaj
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Corresponding author: Anubha Bajaj, MD. (Pathology) Panjab University, Department, of Histopathology, A.B. Diagnostics, A-1, Ring Road , Rajouri Garden, New Delhi, 110027, India —
Competing Interests
The authors have declared that no competing interests exist.
Funding
No specific funding statement was provided by the authors.
Data Availability
No data-availability statement was provided by the authors.
Citation:
Preface
Low grade fibromyxoid sarcoma (LGFMS) is an exceptional, low grade, soft tissue sarcoma with indolent biological behaviour, an extensive preclinical stage, localized tumour reoccurrence and delayed, distant metastasis. The deceptively benign neoplasm was initially scripted by Evans in 1987 and is additionally nomenclated as “Evan’s tumour”. Incidence of sarcomas is nearly 1% of adult malignancies wherein low grade fibromyxoid sarcoma represents beneath <5% of soft tissue sarcomas 1.
Neoplasms configuring collagen rosettes were initially designated as “hyalinising spindle cell tumour with giant rosettes”. Nevertheless, aforesaid neoplasms represent a morphological continuum with low grade fibromyxoid sarcoma and are associated with benign biological behaviour despite manifesting localized tumour reoccurrence or distant metastasis. The minimally cellular sarcoma exhibits a whorled tumour architecture, along with fibrous and myxoid zones composed of uniform, fibroblastic cells with an interposition of curvilinear or arcuate vascular articulations. On account of bland microscopic appearance, the tumefaction may be misinterpreted as a benign mesenchymal neoplasm or a low grade sarcoma. Characteristically, the neoplasms comprehensively (≈100%) harbour a fusion of FUS/CREB3L2 gene 2. Low grade fibromyxoid sarcoma may be concurrent to sclerosing epithelioid fibrosarcoma due to identical chromosomal translocations, common histological features and immune reactivity to mucin 4 (MUC4) 2.
Disease Characteristics
Low grade fibromyxoid sarcoma commonly arises in young adults although middle aged or older individuals within late third to fourth decade may be incriminated. However, around 13% to 19% tumefaction occur below < 18 years and the neoplasm has been documented at 22 months. Age of disease occurrence varies from 6 years to 73 years with a median of 41.8 years. An equivalent gender predisposition is denominated 2, 3.
Low grade fibromyxoid sarcoma is commonly distributed within deep-seated soft tissues of proximal upper or lower extremities and trunk wall. Tumefaction is often located within the thigh, posterior cervical spine, cutaneous surfaces, facial region, mandible, larynx, thoracic cavity, abdomen, bowel and perineum whereas lesions are exceptional in the head and neck, mediastinum, retroperitoneum, colon or perianal space 2, 3.
Low grade fibromyxoid sarcoma demonstrates a superior prognosis although localized tumour reoccurrence and delayed, distant metastasis is observed. Distant metastasis within pulmonary parenchyma, pleura, chest wall and occasionally pericardium, bones, heart or hepatic parenchyma are discerned. On account of indolent clinical behaviour, distant metastasis may appear up to 45 years following initial tumour detection with median detection at 5 years 2, 3.
Localized tumour reoccurrence is observed in around 64% instances, distant metastasis in nearly 45% individuals and disease - related mortality ensues in roughly 42% subjects 2, 3.
On molecular analysis, low grade fibromyxoid sarcoma characteristically displays genomic translocations of chromosome 7 and 16 with emergence of consequent fusion product FUS/ CREB3L2. Fused in sarcoma (FUS)-cyclic AMP responsive element binding protein3-like2 (CREB3L2) is frequently observed in around 75% to 95% of neoplasms. Thus, FUS/CREB3L2 genetic fusion or chromosomal translocation t (7;16)(q32-34;p11) is discerned in a majority (≈90%) of tumefaction with consequent fusion of FUS RNA binding protein. Additionally, FUS/ CREB3L1 genomic fusion is observed in nearly 5% neoplasms along with t(11;16)(p11;p11) genetic translocation. CREB3L2 and CREB2L1 genes encode transcription factors. Nevertheless, concurrence between CREB3L2 fusion gene and localized tumour relapse or metastatic disease is absent 2, 3.
Histological Elucidation
Macroscopically,the neoplasm is gradually progressive, painless, firm, tan coloured, well circumscribed, grossly infiltrative, soft tissue mass of magnitude varying from one centimetre to 18 centimetres, commonly of 6 centimetres diameter. Cut surface is fibrotic with focal myxoid areas4. On fine needle aspiration cytology, the cellular neoplasm is configured by spindle-shaped cells. Tumour cells contain scant, wispy cytoplasm, uniform, elongated nuclei and miniature, inconspicuous nucleoli. The background is prominently myxoid. Significant nuclear pleomorphism or mitotic activity is absent. However, cogent diagnosis on cytology may be challenging 4.5. On low power, the well demarcated neoplasm is composed of alternating foci of myxoid and fibrous tissue. Giant rosettes are intermingled with zones of whorled cellular aggregates recapitulating preliminary rosettes.
Bland tumour cells depict monotonous, hyperchromatic nuclei. Tumour areas can exhibit prominent curvilinear, arching or plexiform vasculature. Mitotic activity is exceptional 4, 5.
Typically, low grade fibromyxoid sarcoma is comprised of a whorled cellular pattern with intermingled fibrous and myxoid areas. Also, minimally cellular areas of collagenous tissue are denominated, composed of uniform spindle-shaped cells4, 5. Unique morphologic pattern of hyalinising spindle cell tumour configuring giant rosettes can appear in certain low grade fibromyxoid sarcomas4, 5.
The minimally to moderately cellular neoplasm is composed of bland, fusiform or spindle-shaped cells with scarce cytoplasm and angulated nuclei. Also, whorled cellular aggregates are exhibited. Focal to diffuse cellular whorls are intermingled within a dense, collagen-rich stroma. Abrupt, focal transition to myxoid areas can be discerned. Roughly 45% neoplasms depict foci of epithelioid cells. Nearly 40% tumours display enlarged, inadequately configured collagen rosettes2, 4.
The neoplasm frequently infiltrates abutting skeletal muscle. Occasionally, tumefaction denominates hyper-cellular foci, tumour necrosis, cellular or nuclear atypia and mitotic figures, features which are characteristic of intermediate to high grade sarcoma. Localized tumour reoccurrence may be hyper-cellular with enhanced mitotic activity 4, 5. On ultrastructural examination, fibroblastic differentiation is exemplified 5. Figure 1, Figure 2, Figure 3, Figure 4, Figure 5, Figure 6, Figure 7, Figure 8.
Figure 1. Low grade fibromyxoid sarcoma enunciating fibrous and myxoid zones of uniform spindle-shaped tumour cells with absence of atypia 9.
Download figure
Figure 2. Low grade fibromyxoid sarcoma exemplifying intermingled foci of fibrous and myxoid regions of spindle-shaped cells with minimal pleomorphism and absence of mitosis (10).
Download figure
Figure 3. Low grade fibromyxoid sarcoma demonstrating alternating fibrous and myxoid areas comprised of uniform spindle-shaped cells (11).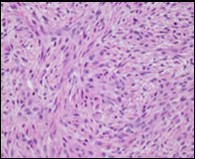
Download figure
Figure 4. Low grade fibromyxoid sarcoma composed of alternating foci of fibrous and myxoid region with interwoven fascicles and bland spindle-shaped tumour cells (12).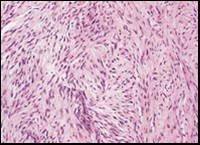
Download figure
Figure 5. Low grade fibromyxoid sarcoma exhibiting alternating fibrous and myxoid zones composed of spindle-shaped tumour cells (13).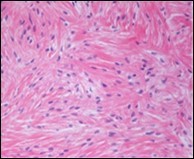
Download figure
Figure 6. Low grade fibromyxoid sarcoma immune reactive to MUC4(14).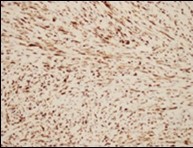
Download figure
Figure 7. Low grade fibromyxoid sarcoma depicting alternating fibrous and myxoid regions with bland, spindle-shaped tumour cells and absent mitosis (15).
Download figure
Figure 8. Low grade fibromyxoid sarcoma delineating alternating fibrous and myxoid areas of minimal cellularity and mitosis (9).
Download figure
Immune Histochemical Elucidation
Assessment of precise immune reactivity is beneficial although staining profile of the benign neoplasm appears nonspecific.
High molecular weight glycoprotein, mucin 4(MUC4) is a sensitive, specific, contemporary immune marker to ascertain and distinguish low grade fibromyxoid sarcoma from histologically identical neoplasms.
A comprehensive (100%), intense, diffuse cytoplasmic immune staining is discerned with MUC4. Gene expression profiling can also be adopted to enunciate MUC4.
Spindle cell neoplasms such as desmoid tumour or soft tissue perineurioma mandating distinction from low grade fibromyxoid sarcoma are immune non reactive to MUC46, 7.
Nearly 25% neoplasms are focally immune reactive to epithelial membrane antigen (EMA) and around 50% tumour nuclei can be diffusely immune stained with p63.
The tumefaction is immune reactive to CD99 (90%), B cell lymphoma (Bcl-2) antigen (90%) or vimentin6, 7.
Epithelial membrane antigen (EMA) is a poorly specific immune marker for discerning low grade fibromyxoid sarcoma although soft tissue perineurioma and a subset of solitary fibrous tumours (SFTs) are immune reactive6, 7.
Low grade fibromyxoid sarcoma is immune non reactive to S100 protein, p40, CD34, nuclear β- catenin, cytokeratin, muscle specific actin (MSA), smooth muscle actin (SMA), SOX10, desmin, h-caldesmon, CD117, murine double minute 2(MDM2) and delay of germination 1(DOG1) antigen Specifically, the delta Np63 isoform of p63 (p40) is consistently immune non reactive6, 7.
Differential Diagnosis
Low grade fibromyxoid sarcoma requires a segregation from associated neoplasms immune reactive to MUC4, denominated by synovial sarcoma (30% to 90%) and sclerosing epithelioid fibrosarcoma (SEF)(78%)3, 4.
Synovial sarcoma is a biphasic neoplasm with distinctive morphology and is composed of spindle-shaped cells and gland-like, epithelial structures, intraluminal mucin, hyper- cellular foci of spheroidal cells with hyperchromatic nuclei, tumour necrosis, frequent mitosis, aggregates of mast cells and focal calcification.
Sclerosing epithelioid fibrosarcoma can be challenging to differentiate as a subset of neoplasms demonstrate fused in sarcoma (FUS) chromosomal translocations. EWSR1/ CREB3L1 genetic fusions are common in sclerosing epithelioid fibrosarcoma (SEF) along with a morphological continuum with low grade fibromyxoid sarcoma and frequent immune reactivity to MUC4. Also, low grade fibromyxoid sarcoma can demonstrate lack of FUS genomic rearrangement with an accompanying EWSR1/CREB3L1 chromosomal fusion3, 4.
Spindle cell neoplasms of head and neck are predominantly (70%) comprised of spindle cell squamous cell carcinoma. “Sarcomatoid” spindle cell squamous cell carcinoma (SCSCC) is discerned in elderly population and demonstrates deceptively bland tumour cells occasionally admixed with collagenous or myxoid matrix. Focal cellular and nuclear pleomorphism with elevated mitotic activity is observed. Spindle cell squamous cell carcinoma is accompanied by dysplasia or malignant metamorphoses of superimposed squamous epithelial layer 3, 4.
Instances depicting an absence of epithelial component can be diagnosed with pertinent immune markers. Around 70% of spindle cell squamous cell carcinomas (SCSCC) are immune reactive to epithelial markers such as cytokeratin AE1/AE3, CK5/6, epithelial membrane antigen (EMA), p63 or p40. A subset also enunciate mesenchymal markers such as CD99, B cell lymphoma 2 (bcl-2), S100 protein and smooth muscle actin (SMA). Generally, immune reactivity to p63 and /or epithelial membrane antigen (EMA) is indicative of spindle cell squamous cell carcinoma 3, 4.
Peripheral nerve sheath tumours as schwannoma or neurofibroma depict alternate, pale, minimally cohesive foci within a spindle cell neoplasm in the absence of overt malignant cytological features. Aforesaid neoplasms exhibit immune reactivity to S100 protein and SOX10 2, 4.
Myxoid neurofibroma is comprised of thick, dense collagen bundles. Tumour cells are incorporated with wavy nuclei. The neoplasm is immune reactive to S100 protein2, 4.
Segregation is also mandated from myofibroblastic neoplasia and fibromatosis.
Fibromatosis constituted by fibroblastic cells configure broad, sweeping fascicles and aligned tumour cells simulate reactive fibroblasts. Definitive, ectatic vascular articulations are observed. Fibromatosis delineates prominent vascular articulations which are distinct from vascular arcades of low grade fibromyxoid sarcoma. Fibromatosis is usually devoid of myxoid areas although occasional myxoid zones may be discerned. Diffuse or occasionally intense, focal nuclear immune reactivity to β catenin is delineated2, 4.
Myofibroblastic neoplasia such as myofibroma or inflammatory myofibroblastic tumour (IMT) are immune reactive to various actins 2, 4.
Fibrosarcoma demonstrates a distinctive fascicular, herringbone tumour configuration and is devoid of myxoid component. Fibrosarcoma is a cautiously arrived at diagnosis of exclusion2, 4.
Myxofibrosarcoma is a predominantly myxoid neoplasm with decimated quantities of fibrous tissue. Significant nuclear pleomorphism and hyperchromasia is observed 2, 4.
Nodular fasciitis is a neoplasm which can be morphologically replicated on tissue culture. Red cell extravasation is exhibited along with foci of myxoid, cystic degeneration2, 4.
Investigative Assay
Low grade fibromyxoid sarcoma can be adequately discerned with the employment of MUC4 immune stain and genomic rearrangement of fused in sarcoma (FUS) gene. Neoplasms depicting a classical morphology which are immune non reactive to MUC4 mandate a cogent molecular analysis which typically demonstrates a FUS/CREB3L2 genetic rearrangement7, 8.
Upon imaging, a multinodular neoplasm is exhibited with alternating intense and minimally enhanced regions.
Molecular studies demonstrate aforementioned chromosomal translocations characteristic of low grade fibromyxoid sarcoma7, 8.
Pertinent investigations for detecting low grade fibromyxoid sarcoma are chromosomal rearrangement of fused in sarcoma (FUS) gene, discerned by fluorescent in situ hybridization (FISH) technique and chimeric fusion genes FUS/CREB3L2 or FUS/CREB3L1 denominated by reverse transcriptase polymerase chain reaction (RT-PCR)7. Adoption of pertinent immune histochemical panel is critical in circumventing misinterpretation of diverse epithelial and mesenchymal neoplasms and aiding appropriate tumour segregation 6, 7.
Therapeutic Options
The aim of optimal treatment strategy is to maximize oncological control with minimized, organ-specific functional impairment.
Comprehensive surgical extermination of localized neoplasm with tumour -free surgical perimeter is a standardized treatment regimen. An en bloc surgical excision with wide periphery of normal/ uninvolved tissue is efficacious7, 8.
Tumour enucleation is optimal for eliminating functional impairment in site-specific neoplasms as the anal sphincter. Localized tumour reoccurrence can be subjected to repetitive surgical excision. Extermination of oligo-metastatic tumefaction can be adopted in specific instances. Generally, around 92.2% neoplasms are subjected to primary surgical resection and around 13.8% tumours mandate repetitive excision in instances of localized tumour reoccurrence7, 8.
Low grade fibromxyoid sarcoma subjected to surgical eradication depict proportionate localized tumour control of 83% and 79% at 5 years and 15 years respectively. Surgical eradication of pulmonary metastasis can be contemplated8.
The indolent neoplasm is not particularly radio sensitive and systemic therapies may be unsatisfactory. Thus, conventional or adjuvant therapy is not recommended. Radiotherapy can be beneficially adopted to treat incriminated tumour margins, pertinent tumour localization or enhanced tumour magnitude, factors which indicate the emergence of a possible, localized tumour relapse or distant metastasis. Radiotherapy enhances localized tumour control in an infiltrative neoplasm, especially with tumours situated within extremities. Radiotherapy with concomitant chemotherapy is employed for treating locally advanced, inoperable neoplasms7, 8.
Radiofrequency ablation and cryo-ablation are beneficial for managing oligo-metastatic sarcomas. Chemotherapy is adopted where the tumefaction has reoccurred locally or disseminated distantly. First line chemotherapy is associated with 0% response rate and a median progression free survival of 1.84%. Previously discerned, progressive neoplasms can be subjected to trabectedin7, 8.
As the neoplasm is exceptional, information pertaining to prognostic and therapeutic outcomes is limited. Significant morbidity can occur. Also, a paucity of evidence is encountered while dealing with advanced or metastatic disease. Thus, contemporary, conventional, systemic chemotherapies demonstrate limited efficacy 7, 8.
Subjects can be disease free at 12 months or display multiple, miniature pulmonary nodules and enlarged regional lymph nodes. Advanced, reoccurring or metastatic disease is not benefitted with systemic or loco- regional treatment.
Tumefaction with uncertain or tumour- incriminated perimeter denominates reoccurrence which may arise up to 15 years following initial surgical excision with a median duration of 3.5 years.
Distant metastasis may ensue within pulmonary parenchyma, pleura or chest wall for a period of up to 45 years following surgery with a median duration of 5 years8.
Emergence of focal areas of intermediate to high grade sarcoma with pertinent morphological features may not influence prognostic outcomes. However, tumour reappearance with a dedifferentiated, anaplastic or round cell morphology and innumerable mitosis can decimate survival. Superficial neoplasms are frequent, especially in paediatric subjects and are accompanied by a favourable prognosis. Miniature tumefaction beneath < 3.5 centimetre magnitude is accompanied by superior outcomes 7, 8.
Extensive clinical monitoring of the neoplasm is necessitated on account of potential delayed localized tumour reoccurrence or distant metastasis. Despite definitive metastatic potential with tumour metastasis discerned decades after initial surgical resection, cogent survival is possible for several years with metastatic disease7, 8.
References
- 1.Evans H L. (1987) Low grade fibromyxoid sarcoma – a report of two metastasizing neoplasms having a deceptively benign appearance”. , Am J Clin Pathol 88(5), 615-9.
- 2.Ban L K, Tseng A H. (2017) Low grade fibromyxoid sarcoma of the external anal sphincter- a case report”. , World J Surg Oncol 15, 109.
- 3.Cowan M L, Thompson L D. (2016) Low grade fibromyxoid sarcoma of the head and neck – a clinicopathologic series and review of literature” Head Neck Pathol. 10(2), 161-166.
- 4.Mendoza A S, O’Leary M P. (2015) Low grade fibromyxoid sarcoma of the sigmoid colon” Exp Mol Pathol. 98(2), 300-303.
- 5.Konecna J, Liberale G. (2015) Diffuse intra-abdominal low grade fibromyxoid sarcoma with hepatic metastasis – case report and review of the literature”. , Int J Surg Case Rep 14, 40-3.
- 6.Lin G, Doyle L A. (2015) An update on the application of newly described immunohistochemical markers in soft tissue pathology” Arch Pathol Lab Med. 139(1), 106-21.
- 7.Chamberlain F. (2020) Engelmann B et al” Low grade fibromyxoid sarcoma-treatment outcomes and efficacy of chemotherapy” In Vivo. 34(1), 239-245.
Cited by (2)
This article has been cited by 2 scholarly works according to:
Citing Articles:
International Journal of Surgery Case Reports (2025) Crossref Semantic Scholar OpenAlex
Medical Journal of Clinical Trials & Case Studies (2020) OpenAlex