Abstract
Targeting cell metabolism is a therapeutic approach which has been used for the treatment of cancers with high levels of proliferation. Inhibition of metabolic processes in cancer cells has shown synergy with current therapeutic options to reduce refractory disease and relapse. In contrast, chronic lymphocytic leukaemia (CLL) is a disease where expansion of the malignant clone results from a combination of enhanced cell survival coupled with low level proliferation. The purpose of this article is to highlight how further research is needed to determine whether targeting cell metabolism may be a viable therapeutic strategy in this disease. We discuss how lymphocyte doubling time (LDT) remains a robust prognostic indicator used in the current clinical management of CLL, and how recognition of CLL as a proliferative disease has led to a greater understanding of the importance of energy-generating processes in its pathobiology. We summarize what is currently known about normal B cell metabolism and consider whether there is evidence of the Warburg effect in CLL cells. Finally, we speculate on how CLL cells may exploit protective mechanisms such as autophagy during times of metabolic stress and how they might influence or be influenced by metabolic characteristics of the microenvironment.
Author Contributions
Academic Editor: Krzysztof Roszkowski, Department of Radiotherapy, F. Lukaszczyk Oncology Center, Poland.
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2014 Chloe E. Clapham, et al.
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Introduction
Chronic lymphocytic leukaemia (CLL) is a common haemic malignancy predominantly of aging adults affecting males more frequently than females.1 This disease is characterized by the accumulation of CD5+ CD20+ CD23+ B cell lymphocytes in the blood, lymph nodes, liver, spleen and bone marrow.2 CLL has a highly variable clinical course in which aggressive disease is characterised by a high tumour burden or, in cases with a low tumour burden, a progressive increase in blood lymphocytes over a period of months. In contrast, in indolent disease the blood lymphocyte count may remain stable over a period of years.3 The lymphocyte doubling time (LDT) was first described in the mid-eighties for its importance in determining CLL prognosis. Specifically, it was shown that patients with a LDT of less than 12 months had a significantly shorter survival time.4 A number of biological markers have subsequently been identified that can separate patients into prognostic groups. These include IGHV mutational status, expression of CD38 and Zeta-chain-associated protein kinase 70 (ZAP-70) and genetic anomalies such as TP53 mutation/deletion.5, 6, 7 Although these factors give some insight into the pathogenesis of CLL and likely prognosis, their ability to reliably predict disease progression in individual patients is limited. Therefore, the decision to commence treatment is still based upon a “wait and watch” strategy whereby therapeutic intervention is applied only once the tumour burden is sufficiently high to cause symptoms or complications, or if the LDT is less than 6 months.3 Thus, LDT remains an important variable in CLL, both as a prognostic factor and also as a guide to treatment initiation.3
In the past it was thought that this characteristic increase in the LDT was due to failed apoptosis.8 Early studies on whole blood samples revealed that the majority of CLL cells are arrested in G0 of the cell cycle, suggesting that lymphocytosis in this disease did not reflect proliferation of the circulating malignant clone.8, 9 Moreover, it was found that CLL cells had high expression levels of anti-apoptotic proteins such as Mcl-1 and Bcl-2 and low expression of pro-apoptotic proteins such as Bax.10, 11, 12, 13 In particular, high levels of Mcl-1 expression were correlated with resistance to chemotherapeutic agents such as chlorambucil and fludarabine.13 Genetic lesions have also been shown to have an anti-apoptotic effect. For example, deletion of p53 (del17p13) leads to the loss of p53-mediated cell death in CLL.14 This is important because agents such as fludarabine exploit the p53 pathway as a mechanism of inducing death of CLL cells, providing one explanation for the evolution of drug resistant clones and the process of relapse.14, 15
It has since been recognised that proliferation plays a major role in CLL pathogenesis.16 A pivotal study by Messmer, et al. 16 used deuterium to label DNA of newly-formed CLL cells and showed that progressive disease was associated with malignant-cell daily birth rates greater than 0.35% of the entire clone. Other studies comparing CLL patients with healthy control subjects have shown that, although CLL cell turnover is lower than that of normal B cells, the rate at which deuterium-labelled CLL cells disappear from circulation is considerably slower.2, 17 Thus, malignant-cell accumulation in CLL is likely dictated by a combination of cell proliferation and resistance to apoptosis, with the longer life span of CLL cells allowing them to relocate into environments that support their survival and further promote proliferation.
Most studies investigating the pathogenesis of CLL continue to focus on mechanisms that promote cell survival and resistance to apoptosis. Very few studies have investigated cell metabolism as a therapeutic target in CLL.18, 19, 20, 21, 22 Recognition of proliferation as a major contributor to the progression of this disease calls for a re-examination of cell metabolism in CLL cells. This is because a key checkpoint in the decision of cells to progress to S phase and undergo proliferation is metabolic status (Figure 1). This makes sense because cells require sufficient macromolecular building blocks and energy in order to successfully complete the process of dividing into daughter cells.23 In a high energy state where the ratio of ATP to AMP is high, a sensor protein known as AMP-kinase (AMPK) remains in an inactive state. This allows employment of the anabolic processes that allow cell growth and proliferation, as well as facilitating spindle orientation during mitosis.24 However, in a low energy state where the level of AMP rises as ATP is depleted, AMPK becomes phosphorylated and active, and works to control the resources available to the cell.25, 26 Activated AMPK does this by inducing catabolic processes such as glycolysis and autophagy, and also blocking cell-cycle progression regulating the function of cyclins and cyclin dependant kinases (CDKs).25, 27 In normal cells, metabolic control of proliferation is influenced by growth factors that stimulate nutrient intake, whereas in malignant cells the role of growth factors can be over-ridden by oncogenic mutations in genes controlling cellular growth.
Figure 1.The role of activated AMPK in regulating the cell cycle.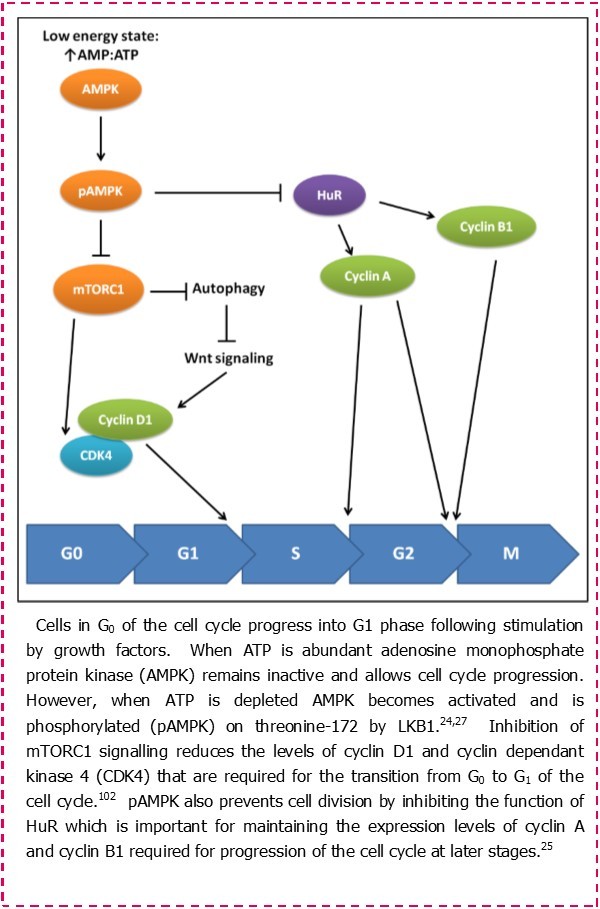
Clearly, there have been many advances in our understanding of CLL since the first studies of CLL cell metabolism in the late 1950s. For example, it was recognised that somatic hypermutation of IGHV genes coding for the B cell receptor (BCR) on CLL cells could be used to differentiate between patients who were likely to have progressive disease; CLL patients whose malignant cells bear unmutated IGHV genes (UM-CLL) are more likely to have progressive disease than those having mutated IGHV genes (M-CLL).5, 28 Analysis of gene expression in UM- and M-CLL cells lead to the discovery that expression of ZAP-70 in CLL cells was connected with poor disease prognosis.29, 30 These observations have led to the development of a hypothesis suggesting that BCR signalling is a major contributor to CLL pathogenesis, particularly with respect to aggressive disease.31 Other factors associated with progressive disease include CD38 expression and genetic abnormalities leading to inactivation of ATM and p53.5, 6, 7 Furthermore, mutation in genes such as SF3B1, NOTCH1 and XPO1 have also been correlated with progressive disease.14, 32, 33, 34, 35, 36, 37 Finally, it is recognised that the pseudofollicles that form in patients with CLL provide an environment that is particularly adept at promoting malignant cell survival and proliferation.11, 38 These factors, either alone or in combination, are likely to determine the precise pattern of behaviour of individual CLL clones.
This article will consider what is known about CLL cell metabolism and examine whether this knowledge needs to be updated in light of new understanding of CLL pathobiology. The therapeutic potential of targeting metabolism in CLL will then be assessed in the light of this improved understanding.
Normal B-Cell Versus CLL-Cell Metabolism
In order to contextualise the metabolic characteristics of CLL cells, an understanding of bioenergetics in normal B cells is needed. Some insight into this process is provided in a recent study by Garcia-Manteiga, et al. 39 who have examined the metabolic profile of B cells as they undergo a transition from resting naive lymphocytes into immunoglobulin-secreting plasma cells. Figure 2 illustrates these changes. Naïve/resting B cells are shown to rely primarily on oxidative phosphorylation for energy production. However, following exposure to antigen these cells begin to utilize aerobic glycolysis as a primary energy source as they become activated and then transit through the germinal centre. While the use of aerobic glycolysis as a metabolic adaptation has reduced efficiency for ATP generation, it confers numerous benefits including a rapid production of the metabolic intermediates and ATP that are required by activated splenic B cells to meet the demands of cell replication.40 A similar process has been described for T cells undergoing activation via the T cell receptor (TCR) and CD28, suggesting a common mechanism of lymphocyte response to antigen receptor stimulation. Importantly, following depletion of glucose stores, T cells further adapt their metabolism by promoting entry of glutamine into the Tricarboxylic acid cycle (TCA cycle) in a process known as glutaminolysis.41 This is likely also to occur in B cells within the splenic environment since increased glutaminolysis prevails as the source of energy production in antibody-producing plasma cells (Figure 2).39 Activated B cells can also differentiate to memory B cells. These cells assume a resting phenotype similar to that associated with naïve B cells, and they regain dependence on oxidative phosphorylation as the primary means of energy generation.
Whether or not CLL cells undergo the same metabolic changes experienced by differentiating normal B cells has not been investigated. Examination of the surface membrane phenotype of CLL cells has shown them to express markers of activation and differentiation, suggesting they resemble antigen-experienced B cells.42 The work by Garcia-Manteiga, et al. 39 on B-cell bioenergetics suggests that circulating CLL cells may rely on oxidative phosphorylation as their primary means of energy generation owing to their resting state. This notion is supported by Moran, et al. 43 who showed that high levels of oxidative stress experienced by CLL cells in the circulation is the result of aerobic mitochondrial respiration. However, a recent study of metabolites in serum samples from CLL patients shows increased levels of pyruvate and glutamate compared to serum samples from normal participants.19 Further analysis comparing the metabolic profile of serum samples between UM-CLL and M-CLL patients revealed that increased lactate, fumarate and uridine levels were associated with UM-CLL patients indicating that UM-CLL cells have a greater reliance on glycolysis. While this study did not directly examine CLL cell metabolism, the implications are that the changes in metabolic profile observed between CLL cells and normal cells are caused by metabolic reprogramming of CLL cells.19
Figure 2.Metabolic adaptations following the activation of normal B cells.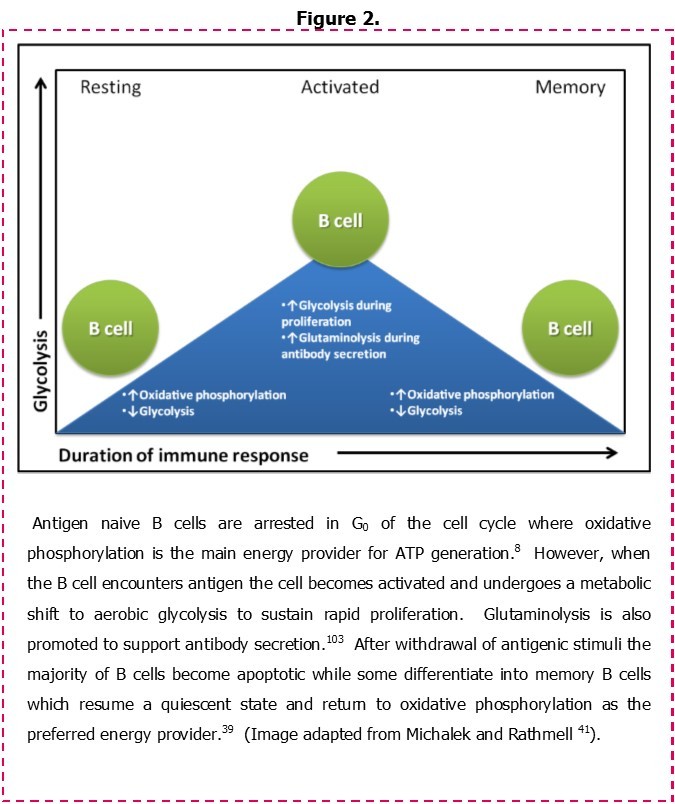
The Warburg Effect: Is Metabolic Deregulation Occurring in CLL Cells?
Reprogramming of key metabolic mediators is characteristic of the Warburg effect, a phenomenon whereby malignant cells upregulate glycolytic activity in the presence of oxygen and rapidly consume glucose.44 That UM-CLL cells may have greater reliance on glycolysis for energy production19 agrees with the model of aerobic glycolysis induction following BCR stimulation in normal B cells.39 This is supported by an existing paradigm whereby UM-CLL cells experience constitutive in-vivo signalling through the BCR45, 46, and from studies demonstrating that UM-CLL cells derive from pre-germinal centre CD5+ B cells whereas M-CLL cells derive from post-germinal centre CD5+, CD27+ B cells.47 BCR stimulated induction of glycolysis is demonstrated to involve protein kinase Cβ (PKCβ)48, and CLL cells may be particularly sensitive because they overexpress PKCβII.49 That metabolic reprogramming is a feature of CLL cells comes from a study of sucrose isomaltase function in relation to mutation. Sucrose isomaltase is the fifth most mutated gene in CLL, and mutations resulting in loss of enzyme function are associated with increased expression of genes associated with glucose metabolism.50 Furthermore, Tili, et al. 51 have correlated metabolic reprogramming with poor prognosis and have demonstrated that reduced expression of the microRNA (miR) miR-125b in CLL cells leads to increased expression of enzymes involved in glucose metabolism, potentially explaining observed increased levels of glycolytic intermediates in plasma and urine of CLL patients with progressive disease. In addition to this, microRNA’s known to be deregulated in CLL have been shown to play an important role modulating metabolic pathways in other cell types.52, 53, 54, 55 For instance, two of the most important microRNA’s in relation to CLL pathogenesis; miR-15a, miR-16 have been implicated in the regulation of ATP generating processes as has another microRNA; miR-195.52 Other microRNA’s associated with mitochondrial function such as miR-23a/b, miR-126, and miR-155 have also been shown to mediate glucose metabolism.52, 53, 56, 57 However, early studies examining glucose uptake by CLL cells show that they consume less glucose than do normal B lymphocytes.8 Studies using fluorodeoxyglucose positron emission tomography (FDG-PET) to visualise CLL cells in vivo have met with poor results; sensitivity of detection was 53% and the extent of disease was often underestimated.58 This may be because CLL is composed of malignant cell fractions with different proliferative activities whereby recently divided cells and older/quiescent cells may have different glucose requirements. This notion is supported by a report indicating that CLL patients have two populations of circulating malignant cells with different degrees of mitochondrial polarisation and dependencies on glucose.59 Where FDG-PET has been effectively used in CLL management is with respect to the detection of Richter’s transformation.60 In this situation CLL cells change into faster growing diffuse large B cell lymphoma (DLBCL) or Hodgkin’s lymphoma cells and likely gain independence from the microenvironmental cues that normally regulate their proliferation. The increased need of glucose by these cells may be due to activation of the Jak/STAT and PI3K/Akt/mTOR signalling pathways as has been suggested by a study comparing metabolic profiles between activated T cell lymphocytes and lymphoma cells.61
While the level of glucose uptake experienced by CLL cells appears to be at odds with the high glucose consumption that is characteristic of the Warburg effect, the glycolytic pathway is nevertheless important to CLL cell pathophysiology. This is because treatment of CLL cells with the hexokinase inhibitor lonidamine has been shown to result in decreased cell viability in vitro and in reductions in lymphocyte count as well as in the size of the lymph nodes and spleen when it is used in vivo.18 Furthermore, a more recent study by Tidmarsh, et al. 62 has shown that inhibition of the glycolytic pathway in CLL cells using 2-deoxyglucose (2DG) results in a reduction of ATP levels followed by a loss of cell viability. The lower level of glucose uptake in CLL cells may reflect the use of an alternative source of glucose. A possible candidate of this alternative source is glycogen which has been shown to accumulate in CLL cells63, negating the need for glucose uptake. However, this simple explanation is complicated by observations that glycogen phosphorylase activity in PHA-activated CLL cells is lower than that in normal lymphocytes64, indicating that CLL cells may have reduced capability of converting glycogen to glucose. Alternatively hypoxia inducible factor 1α (HIF-1α) may be responsible for increased glycogen storage as it is reported to do in other cancerous and non-cancerous cells.65 Expression of HIF-1α is raised in circulating CLL cells due to down regulation of von Hippel-Lindau (pVHL) protein which catalyses HIF-1a degradation.66 Importantly, studies of glucose uptake/usage by CLL cells have mainly been performed on cultured cells. Cultured CLL cells, like those in circulation are mainly in G0, and their requirement for glucose may be very low. These cells may rely on oxidative phosphorylation as we have already discussed, explaining why drugs such as lonidamine and 2DG have their cytotoxic effects. Finally, CLL cells may utilize fatty acid oxidation as a source of glucose as has been suggested in a recent paper by Spaner, et al. 22. Here they demonstrate that PPARα, a mediator of fatty acid oxidation, is upregulated in circulating CLL cells, and that CLL cells show in vitro and in vivo sensitivity to a specific inhibitor of PPARα, MK886.
Coping in Times of Metabolic Stress: Autophagy and Metabolic Adaptations to Quiescence in CLL Cells
CLL cells survive in the circulation for a considerably longer time than do normal B cells.2 In this regard the longer a CLL cell clone can survive in circulation, the better chance it will have of re-entering tissues where it will receive signals for proliferation and enhanced survival. The prolonged lifespan of CLL cells in circulation likely results from a combination of apoptotic resistance mechanisms and metabolic adaptations that help to maintain viability.65 One such metabolic adaptation may result from the increased expression of HIF-1α which may maintain CLL cells in an arrested state and hold them from progressing to cellular senescence by inhibiting mammalian target of rapamycin (mTOR) (Figure 3).67, 68, 69 This HIF-1a-induced adoption of a quiescent phenotype may explain why early studies showed that CLL cells consume less glucose than do normal B lymphocytes8, and why CLL cells are not easily imaged by FDG-PET.58 Moreover, adoption of a quiescent phenotype may be related to progressive disease in CLL because malignant cell susceptibility to apoptosis, low level RNA content and high expression of p27kip1 all correlate to patients with late stage disease.67, 70, 71 This relationship is supported by further observations showing that UM-CLL and ZAP-70 expression in CLL cells also correlate with ability to adopt a quiescent phenotype.72
Figure 3.Regulation of cell metabolism via mTORC1 and mTORC2 following BCR and CD40 stimulation.
Another consequence of mTOR inhibition is the induction of autophagy, a self-digestive process initiated in cells experiencing metabolic stress. During autophagy catabolism of internal organelles is activated to generate the ATP that is necessary to maintain essential housekeeping functions and cell survival.69, 73 CLL cells are reported to experience autophagy; one early study has suggested that as much as one third of the overall protein degradation that takes place in these cells is due to this process74, while more recent studies have suggested that IL-24 induces its pro-survival effect by inducing autophagy in CLL cells.75 A study by Mahoney, et al. 76 has also shown CLL cells to express the ATG family of proteins critical for autophagy in addition to demonstrating autophagy induction by known agents. Furthermore, our own unpublished observations indicate that a major regulator of mTOR, AMPK, is constitutively phosphorylated on the serine residues associated with enzyme activation, and that phospho-AMPK levels seem to be increased in UM-CLL compared to M-CLL cells. This observation agrees with a published result showing that an activator of AMPK, sirtuin 1 (SIRT1), is upregulated in CLL cells.77 That autophagy in CLL cells has a cytoprotective effect is recently demonstrated in a study showing that re-expression of the miRNA gene miR-130a inhibits autophagy in CLL cells and sensitizes them to apoptosis.78 Moreover, a further study by Maccallum, et al. 79 has shown similar re-sensitization to apoptosis when autophagy in CLL cells is disrupted by the SIRT1 inhibitor Tenovin-6.
Excessive stimulation of autophagy can also result in apoptosis through a process called autophagic cell death (ACD).80 In this process organelle digestion continues beyond the point that can be restored when metabolic stress is removed. ACD is observed in CLL cells that have been treated with the AMP analogue acadesine (AICAR), due to the ability of this compound to activate AMPK and inhibit mTOR function.81 A study by Santidrian, et al. 82 has shown this effect can occur independently of AMPK and p53 in CLL cells via the Bcl-2 family proteins; BIM, NOXA and PUMA. Further to this, the death inducing effects of the glucocorticoid dexamethasone has been reported to arise due to the stimulation of autophagy in CLL cells.80, 83 Notably, UM-CLL cells have been found to be significantly more sensitive to dexamethasone-induced killing in keeping with the observations that these cells have greater levels of AMPK activation and may be more readily pushed over the edge to ACD.84 A further study by Mahoney, et al. 76 describes how other select chemotherapeutic agents such as fludarabine, the PI3K δ-isoform inhibitor CAL-101, thapsigargin and flavopiridol all stimulate the induction of autophagy prior to inducing apoptotic cell death. However, the cytotoxic effect of thapsigargin and flavopiridol can be enhanced following disruption of ER stress-induced autophagy, suggesting that autophagy induced through this mechanism is protective against cell death and promotes drug resistance. This mechanism of autophagy has been associated with dasatinib resistance in CLL patients.85
Although autophagy has been studied extensively from the perspective of protection from metabolic stress in circulating cells, it has also been shown to have a role within the microenvironment. Recycling of nutrients may occur not only within the cancerous cells themselves but also in the adjacent stroma as a means of providing nutrients to promote invasion and metastasis.86, 87 A possible mechanism of autophagy induction and metabolic reprogramming in adjacent stromal cells is through the generation of oxidative stress76 as has been observed in the tumour microenvironment of breast cancer.88 This study showed that the production of reactive oxygen species (ROS) by breast cancer cells causes the bordering fibroblasts to switch to glycolysis and generate fuel for the breast cancer cells in the form of pyruvate, which then feeds into oxidative phosphorylation. This synergistic relationship is called the reverse Warburg effect because of the induction of the abnormal glycolytic phenotype in the supporting fibroblasts as opposed to the malignant breast cancer cells. This symbiosis is suggested to occur via shuttling of metabolic intermediates such as lactate and pyruvate through monocarboxylate transporters (MCT) -1 and -4 on cancerous cells and the cells of the microenvironment.89, 90
Microenvironmental Influences on CLL Cell Bioenergetics
The relationship between the microenvironment and CLL cells is important, particularly in the context of apoptotic resistance and induction of cell proliferation. The interaction of quiescent CLL cells with the microenvironment has been modelled using supportive cells such as bone marrow stromal cells, endothelial cells and parental fibroblasts as well as fibroblasts which express CD40 ligand (CD40L) to simulate the influence of T helper cells on CLL cell proliferation. Soluble factors such as IL-21 and IL-4 may also be added to the system to replicate cytokines secreted by T cells.91 Thus, these co-culture systems are thought to mirror the interactions which occur in vivo to induce cell activation, division and enhanced survival in CLL.92 What is missing is an assessment of the impact these interactions have on the metabolism of these cells. Existing studies of these interactions may be useful in directing future research. For example, a study by Willimott and Wagner 93 describes changes in the profile of microRNA’s (miR’s) expressed in CLL cells following co-culture with parental and CD40L-expressing fibroblasts. This study identified multiple miR’s that were deregulated in CLL cells cultured on fibroblasts and a cursory examination of miR’s that potentially regulate metabolic pathways can be observed. In particular, incubation of CLL cells with parental fibroblasts induces changes in the expression of miR-125b (whose effects we discuss in a previous section of this review) along with let-7c which is known to regulate glucose metabolism as part of the lin28/let-7 pathway.94 Furthermore, incubation with CD40L-expressing fibroblasts induces increased expression of miR-155, which can function in regulating glycolytic activity.95 The effect of CD40L on CLL cells may be akin to that observed on similarly stimulated endothelial cells where activation of mTOR (Figure 3) and an increase in cell size has been demonstrated.96 Our own unpublished experiments of CLL cells co-cultured with CD40L-expressing fibroblasts show increased cell size providing support for this direction of endeavour. Moreover, a phase 2 clinical trial testing the mTOR inhibitor everolimus shows that this compound can stimulate mobilization of CLL cells from nodal tissues into the circulation, suggesting that the mTOR pathway may be important for maintaining CLL cells within the microenvironment.97 With respect to cytokines, IL-4 has been shown to have a role in the regulation of glucose levels in normal B cells98, but this remains unexamined in CLL cells. Finally, interaction with adhesion molecules such as E-Cadherin could possibly regulate metabolic processes through the induction of HIF-1α expression as has been demonstrated for breast cancer cells.99
CLL cells may also influence their microenvironment and reprogram it to provide protection from oxidative stress and clearance by the immune system. A recent study by Lutzny, et al. 100 showed that CLL cell contact with stromal cells induced expression of PKCβII in these cells which resulted in activation of the NFκB pathway and secretion of growth factors that, in turn, enhanced the survival of the CLL cells. Zhang, et al. 101 has also demonstrated how CLL cell survival is enhanced by modifications to the supportive cells. In this study bone marrow stromal cells are shown to supply the metabolic intermediate cysteine to CLL cells. This is important for CLL cell survival because this amino acid is required for the production of glutathione (GSH), a redox mediator which prevents the accumulation of ROS.
Conclusions
The purpose of this manuscript is to highlight the need for further research to assess whether targeting cell metabolism may be a viable strategy for the treatment of CLL, a disease with a low level of proliferative activity. While some studies have successfully targeted metabolism in CLL the metabolic pathways used by these cells have not been characterized. As discussed above CLL cells exist in two states which will have different metabolic requirements and local resources. In the peripheral circulation CLL cells may use oxidative phosphorylation similar to their normal counterparts or on the other hand these cells may use an alternative source of glucose such as fatty acid oxidation. There is also evidence which implies metabolism is slowed as the cells enter a quiescent state and that mechanisms which provide protection from metabolic stress such as hypoxia and autophagy are exploited. Importantly, there is evidence which correlates poor prognostic indicators with the ability of these cells to withstand nutrient deprivation suggesting that CLL cells are more likely to survive in the circulation and re-enter the haemic tissues. Furthermore, there is evidence of metabolic reprogramming occurring in CLL cells correlated with bad disease suggesting that these cells are more able to change their metabolic phenotype to meet energy demands. During the proliferative stage located in the secondary tissues, CLL cells likely undergo a metabolic shift to glycolysis to support proliferative activity. In order to sustain proliferation, CLL cells may influence the metabolism of the surrounding cells in the microenvironment to provide substrates and intermediates taking advantage of mechanisms which protect from oxidative stress and promote autophagy. Taken together these findings emphasize the importance of further research to better understand the difference in CLL cell metabolism between these two states and how this may provide key insights for the development of new treatments for this disease.
Acknowledgements
This work was supported by AstraZeneca (Manchester, UK.)
References
- 2.Defoiche J, Debacq C, Asquith B. (2008) Reduction of B cell turnover in chronic lymphocytic leukaemia. , British Journal of Haematology 143(2), 240-247.
- 3.Oscier D, Fegan C, Hillmen P. (2004) Guidelines on the diagnosis and management of chronic lymphocytic leukaemia. , British Journal of Haematology 125(3), 294-317.
- 4.Montserrat E, Sanchezbisono J, Vinolas N, Rozman C. (1986) Lymphocyte doubling time in chronic lymphocytic leukemia - analysis of its prognostic signifcance. , British Journal of Haematology 62(3), 567-575.
- 5.Damle R N, Wasil T, Fais F. (1999) Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. , Blood 94(6), 1840-1847.
- 6.Gonzalez D, Martinez P, Wade R. (2011) Mutational status of the TP53 gene as a predictor of response and survival in patients with chronic lymphocytic leukemia: results from the LRF CLL4 trial. , Journal of Clinical Oncology 29(16), 2223-2229.
- 7.Crespo M, Bosch F, Villamor N. (2003) ZAP-70 expression as a surrogate for immunoglobulin-variable-region mutations in chronic lymphocytic leukemia. , New England Journal of Medicine 348(18), 1764-1775.
- 8.Brody J I, Oski F A, Singer D E. (1969) Impaired pentose phosphate shunt and decreased glycolytic activity in lymphocytes of chronic lymphocytic leukemia metabolic pathway. , Blood-the Journal of Hematology 34(4), 421-429.
- 9.Dameshek W. (1967) Chronic lymphocytic leukemia-an accumulative disease of immunolgically incompetent lymphocytes. , Blood 29(4), 566-584.
- 10.Gottardi D, Alfarano A, De Leo AM, Stacchini A, Bergui L et al. (1995) Defective apoptosis due to bcl-2 overexpression may explain why B-CLL cells accumulate in G0. Current topics in microbiology and immunology. 194, 307-312.
- 11.Zenz T, Mertens D, Kueppers R, Doehner H, Stilgenbauer S. (2010) From pathogenesis to treatment of chronic lymphocytic leukaemia. , Nature Reviews Cancer 10(1), 37-50.
- 12.Pepper C, Hoy T, Bentley D P. (1997) Bcl-2/Bax ratios in chronic lymphocytic leukaemia and their correlation with in vitro apoptosis and clinical resistance. , British Journal of Cancer 76(7), 935-938.
- 13.Kitada S, Andersen J, Akar S. (1998) Expression of apoptosis-regulating proteins in chronic lymphocytic leukemia: correlations with in vitro and in vivo chemoresponses. , Blood 91(9), 3379-3389.
- 14.Dohner H, Fischer K, Bentz M. (1995) P53 gene deletion predicts for poor survival and non-response to therapy with purine analogs in chronic B cell leukemias. , Blood 85(6), 1580-1589.
- 15.Pettitt A R, Sherrington P D, Cawley J C. (1999) The effect of p53 dysfunction on purine analogue cytotoxicity in chronic lymphocytic leukaemia. , British Journal of Haematology 106(4), 1049-1051.
- 16.Messmer B T, Messmer D, Allen S L. (2005) In vivo measurements document the dynamic cellular kinetics of chronic lymphocytic leukemia B cells. , Journal of Clinical Investigation 115(3), 755-764.
- 17.R van Gent, Kater A P, Otto S A. (2008) In vivo dynamics of stable chronic lymphocytic leukemia inversely correlate with somatic hypermutation levels and suggest no major leukemic turnover in bone marrow. , Cancer Research 68(24), 10137-10144.
- 18.Tura S, Cavo M, Gobbi M, Franchi P. (1984) Lonidamine in the treatment of chronic lymphocytic leukemia. , Oncology 41, 90-93.
- 19.MacIntyre D A, Jiménez B, Lewintre E J. (2010) Serum metabolome analysis by 1H-NMR reveals differences between chronic lymphocytic leukaemia molecular subgroups. , Leukemia 24(4), 788-797.
- 20.Samudio I, Fiegl M, McQueen T, Clise-Dwyer K, Andreeff M. (2008) The Warburg effect in leukemia-stroma cocultures is mediated by mitochondrial uncoupling associated with uncoupling protein 2 activation. , Cancer Research 68(13), 5198-5205.
- 21.Doughty C A, Bleiman B F, Wagner D J. (2006) Antigen receptor-mediated changes in glucose metabolism in B lymphocytes: role of phosphatidylinositol 3-kinase signaling in the glycolytic control of growth. , Blood 107(11), 4458-4465.
- 22.Spaner D E, Lee E, Shi Y. (2013) PPAR-alpha is a therapeutic target for chronic lymphocytic leukemia. , Leukemia 27(5), 1090-1099.
- 23.Garedew A, Andreassi C, Moncada S. (2012) Mitochondrial dynamics, biogenesis, and function are coordinated with the cell cycle by APC/C-CDH1. Cell Metabolism. 15(4), 466-479.
- 24.Thaiparambil J T, Eggers C M, Marcus A I. (2012) AMPK regulates mitotic spindle orientation through phosphorylation of myosin regulatory light chain. Molecular and Cellular Biology. 32(16), 3203-3217.
- 25.Wang W, Fan J, Yang X. (2002) AMP-activated kinase regulates cytoplasmic HuR. Molecular and Cellular Biology. 22(10), 3425-3436.
- 26.Valcourt J R, JMS Lemons, Haley E M, Kojima M, Demuren O O et al. (2012) Staying alive: metabolic adaptations to quiescence. , Cell Cycle 11(9), 1680-1696.
- 27.Vadlakonda L, Pasupuleti M, Pallu R. (2013) Role of PI3K-AKT-mTOR and Wnt Signaling Pathways. in Transition of G1-S Phase of Cell Cycle in Cancer Cells. Frontiers in oncology.3 85-85.
- 28.Hamblin T J, Davis Z, Gardiner A, Oscier D G, Stevenson F K. (1999) Unmutated Ig V-H genes are associated with a more aggressive form of chronic lymphocytic leukemia. , Blood 94(6), 1848-1854.
- 29.Wiestner A, Rosenwald A, Barry T S. (2003) ZAP-70 expression identifies a chronic lymphocytic leukemia subtype with unmutated immunoglobulin genes, inferior clinical outcome, and distinct gene expression profile. , Blood 101(12), 4944-4951.
- 30.Rosenwald A, Alizadeh A A, Widhopf G. (2001) Relation of gene expression phenotype to immunoglobulin mutation genotype in B cell chronic lymphocytic leukemia. , Journal of Experimental Medicine 194(11), 1639-1647.
- 31.Stevenson F K, Krysov S, Davies A J, Steele A J, Packham G. (2011) B-cell receptor signaling in chronic lymphocytic leukemia. , Blood 118(16), 4313-4320.
- 32.Starostik P, Manshouri T, O'Brien S. (1998) Deficiency of the ATM protein expression defines an aggressive subgroup of B-cell chronic lymphocytic leukemia. , Cancer Research 58(20), 4552-4557.
- 33.Schaffner C, Stilgenbauer S, Rappold G A, Dohner H, Lichter P. (1999) Somatic ATM mutations indicate a pathogenic role of ATM in B-cell chronic lymphocytic leukemia. , Blood 94(2), 748-753.
- 34.Wattel E, Preudhomme C, Hecquet B. (1994) P53 mutations are associated with resistance to chemotherapy and short survival in hematologic malignancies. , Blood 84(9), 3148-3157.
- 35.Rossi D, Bruscaggin A, Spina V. (2011) Mutations of the SF3B1 splicing factor in chronic lymphocytic leukemia: association with progression and fludarabine-refractoriness. , Blood 118(26), 6904-6908.
- 36.Fabbri G, Rasi S, Rossi D. (2011) Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. , Journal of Experimental Medicine 208(7), 1389-1401.
- 37.Puente X S, Pinyol M, Quesada V. (2011) Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. , Nature 475(7354), 101-105.
- 38.Pleyer L, Egle A, Hartmann T N, Greil R. (2009) Molecular and cellular mechanisms of CLL: novel therapeutic approaches. Nature Reviews Clinical Oncology. 6(7), 405-418.
- 39.Garcia-Manteiga J M, Mari S, Godejohann M. (2011) Metabolomics of B to plasma cell differentiation. , Journal of Proteome Research 10(9), 4165-4176.
- 40.Cheung E C, Vousden K H. (2010) The role of p53 in glucose metabolism. , Current Opinion in Cell Biology 22(2), 186-191.
- 41.Michalek R D, Rathmell J C. (2010) The metabolic life and times of a T-cell. Immunological Reviews. 236, 190-202.
- 42.Damle R N, Ghiotto F, Valetto A. (2002) B-cell chronic lymphocytic leukemia cells express a surface membrane phenotype of activated, antigen-experienced B lymphocytes. , Blood 99(11), 4087-4093.
- 43.Moran E C, Kamiguti A S, Cawley J C, Pettitt A R. (2002) Cytoprotective antioxidant activity of serum albumin and autocrine catalase in chronic lymphocytic leukaemia. , British Journal of Haematology 116(2), 316-328.
- 44.Warburg O, Posener K, Negelein E. (1924) On the metabolism of carcinoma cells. , Biochemische Zeitschrift 152, 309-344.
- 45.Chiorazzi N, Ferrarini M. (2003) B cell chronic lymphocytic leukemia: lessons learned from studies of the B cell antigen receptor. Annual Review of Immunology. 21, 841-894.
- 46.Krysov S, Potter K N, Mockridge C I. (2010) Surface IgM of CLL cells displays unusual glycans indicative of engagement of antigen in vivo. , Blood 115(21), 4198-4205.
- 47.Seifert M, Sellmann L, Bloehdorn J. (2012) Cellular origin and pathophysiology of chronic lymphocytic leukemia. , Journal of Experimental Medicine 209(12), 2183-2198.
- 48.Blair D, Dufort F J, Chiles T C. (2012) Protein kinase C beta is critical for the metabolic switch to glycolysis following B-cell antigen receptor engagement. , Biochemical Journal 448, 165-169.
- 49.Abrams S T, Lakum T, Lin K. (2007) B-cell receptor signaling in chronic lymphocytic leukemia cells is regulated by overexpressed active protein kinase C beta II. , Blood 109(3), 1193-1201.
- 50.Rodriguez D, Ramsay A J, Quesada V. (2013) Functional analysis of sucrase-isomaltase mutations from chronic lymphocytic leukemia patients. Human Molecular Genetics. 22(11), 2273-2282.
- 51.Tili E, Michaille J-J, Luo Z.(2012)The down-regulation of miR-125b in chronic lymphocytic leukemias leads to metabolic adaptation of cells to a transformed state. , Blood 120(13), 2631-2638.
- 52.Nishi H, Ono K, Iwanaga Y. (2010) microRNA-15b modulates cellular ATP levels and degenerates mitochondria via Arl2 in neonatal rat cardiac myocytes. , Journal of Biological Chemistry 285(7), 4920-4930.
- 53.Volinia S, Galasso M, Costinean S. (2010) Reprogramming of miRNA networks in cancer and leukemia. , European Journal of Medical Research 15, 152-152.
- 54.Bienertova-Vasku J, Sana J, Slaby O. (2013) The role of microRNAs in mitochondria in cancer. , Cancer Letters 336(1), 1-7.
- 55.Kim H-R, Roe J-S, Lee J-E, Cho E-J, Youn H-D. (2013) p53 regulates glucose metabolism by miR-34a. , Biochemical and Biophysical Research Communications 437(2), 225-231.
- 56.Zhu D-X, Zhu W, Fang C. (2012) miR-181a/b significantly enhances drug sensitivity in chronic lymphocytic leukemia cells via targeting multiple anti-apoptosis genes. , Carcinogenesis 33(7), 1294-1301.
- 57.Zanette D L, Rivadavia F, Molfetta G A. (2007) miRNA expression profiles in chronic lymphocytic and acute lymphocytic leukemia. , Brazilian Journal of Medical and Biological Research 40(11), 1435-1440.
- 58.Karam M, Novak L, Cyriac J, Ali A, Nazeer T et al. (2006) Role of fluorine-18 fluoro-deoxyglucose positron emission tomography scan in the evaluation and follow-up of patients with low-grade lymphomas. , Cancer 107(1), 175-183.
- 59.Gardner J R, Devlin S, Knapp K. (2012) Aerobic glycolysis predicts outcome in early chronic lymphocytic leukemia. , ASH Annual Meeting Abstracts 120(21), 2482.
- 60.Bruzzi J F, Macapinlac H, Tsimberidou A M. (2006) Detection of richter's transformation of chronic lymphocytic leukemia by PET/CT. , Journal of Nuclear Medicine 47(8), 1267-1273.
- 61.Altman B J, Dang C V. (2012) Normal and cancer cell metabolism: lymphocytes and lymphoma. , Febs Journal 279(15), 2598-2609.
- 62.Tidmarsh G F, Tanner L I, O'Connor J, Eng C, Mordec K. (2004) Effect of 2-deoxyglucose, a glycolytic Inhibitor, on the ATP levels and viability of B lymphocytes from subjects with chronic lymphocytic leukemia (CLL). ASH Annual Meeting Abstracts. 104(11), 4810.
- 63.Mitus W J, Bergna L J, Mednicoff I B, Dameshek W.Cytochemical studies of glycogen content of lymphocytes in lymphocytic proliferations. , Blood 13(8), 748-756.
- 64.Monahan T M, Marchand N W, Fritz R R, Abell C W. (1975) Cyclic adenosine 3'-5'-monophosphate levels and activities of related enzymes in normal and leukemic lymphocytes. , Cancer Research 35(9), 2540-2547.
- 65.Pelletier J, Bellot G, Gounon P, Lacas-Gervais S, Pouyssegur J et al. (2012) Glycogen synthesis is induced in hypoxia by the hypoxia-inducible factor and promotes cancer cell survival. Frontiers in oncology. 2, 18.
- 66.Ghosh A K, Shanafelt T D, Cimmino A. (2009) Aberrant regulation of pVHL levels by microRNA promotes the HIF/VEGF axis in. , CLL B cells. Blood 113(22), 5568-5574.
- 67.Ricciardi M R, Petrucci M T, Gregorj C. (2001) Reduced susceptibility to apoptosis correlates with kinetic quiescence in disease progression of chronic lymphocytic leukaemia. , British Journal of Haematology 113(2), 391-399.
- 68.Arsham A M, Howell J J, Simon M C. (2003) A novel hypoxia-inducible factor-independent hypoxic response regulating mammalian target of rapamycin and its targets. , Journal of Biological Chemistry 278(32), 29655-29660.
- 69.Leontieva O V, Natarajan V, Demidenko Z N, Burdelya L G, Gudkov A V et al. (2012) Hypoxia suppresses conversion from proliferative arrest to cellular senescence. Proceedings of the National Academy of Sciences of the United States of America 109(33), 13314-13318.
- 70.Vermes I, Haanen C, Richel D J, Schaafsma M R, KalsbeekBatenburg E et al. (1997) Apoptosis and secondary necrosis of lymphocytes in culture. , Acta Haematologica 98(1), 8-13.
- 71.Consoli U, El-Tounsi I, Sandoval A. (1998) Differential induction of apoptosis by fludarabine monophosphate in leukemic B and normal T cells in chronic lymphocytic leukemia. , Blood 91(5), 1742-1748.
- 72.Danilov A V, Danilova O V, Brown J R, Rabinowitz A, Klein A K et al. (2010) Dipeptidyl peptidase 2 apoptosis assay determines the B-cell activation stage and predicts prognosis in chronic lymphocytic leukemia. Experimental Hematology. 38(12), 1167-1177.
- 73.Marelli-Berg F M, Fu H, Mauro C. (2012) Molecular mechanisms of metabolic reprogramming in proliferating cells: implications for T-cell-mediated immunity. , Immunology 136(4), 363-369.
- 74.Seglen P O, Munthekaas A C, MAS Dybedal. (1984) Amino acid control of protein degradation in normal and leukemic human lymphocytes. , Experimental Cell Research 155(1), 121-128.
- 75.Yang C, Tong Y, Ni W. (2010) Inhibition of autophagy induced by overexpression of mda-7/interleukin-24 strongly augments the antileukemia activity in vitro and in vivo. Cancer Gene Therapy. 17(2), 109-119.
- 76.Mahoney E, Byrd J C, Johnson A J.Autophagy and ER stress play an essential role in the mechanism of action and drug resistance of the cyclin-dependent kinase inhibitor flavopiridol. , Autophagy 9(3), 434-435.
- 77.Audrito V, Vaisitti T, Rossi D. (2011) Nicotinamide blocks proliferation and induces apoptosis of chronic lymphocytic leukemia cells through activation of the p53/miR-34a/SIRT1 tumor suppressor network. , Cancer Research 71(13), 4473-4483.
- 78.Kovaleva V, Mora R, Park Y J. (2012) miRNA-130a targets ATG2B and DICER1 to inhibit autophagy and trigger killing of chronic lymphocytic leukemia cells. , Cancer Research 72(7), 1763-1772.
- 79.Maccallum S F, Groves M J, James J. (2013) Dysregulation of autophagy in chronic lymphocytic leukemia with the small-molecule sirtuin inhibitor tenovin-6. Scientific reports. 3, 1275-1275.
- 80.Laane E, Tamm K P, Buentke E. (2009) Cell death induced by dexamethasone in lymphoid leukemia is mediated through initiation of autophagy. Cell Death and Differentiation. 16(7), 1018-1029.
- 81.Neste E Van Den, Cazin B, Janssens A. (2013) Acadesine for patients with relapsed/refractory chronic lymphocytic leukemia (CLL): a multicenter phase I/II study. Cancer chemotherapy and pharmacology. 71(3), 581-591.
- 82.Santidrian A F, Gonzalez-Girones D M, Iglesias-Serret D.(2010)AICAR induces apoptosis independently of AMPK and p53 through up-regulation of the BH3-only proteins BIM and NOXA in chronic lymphocytic leukemia cells. , Blood 116(16), 3023-3032.
- 83.Grander D, Kharaziha P, Laane E, Pokrovskaja K, Panaretakis T. (2009) Autophagy as the main means of cytotoxicity by glucocorticoids in hematological malignancies. , Autophagy 5(8), 1198-1200.
- 84.Melarangi T, Zhuang J, Lin K. (2012) Glucocorticoid resistance in chronic lymphocytic leukaemia is associated with a failure of upregulated. Bim/Bcl-2 complexes to activate Bax and Bak. Cell Death and Disease 3, 327.
- 85.Amrein L, Soulieres D, Johnston J B, Aloyz R. (2011) p53 and autophagy contribute to dasatinib resistance in primary CLL lymphocytes. , Leukemia Research 35(1), 99-102.
- 86.Bonuccelli G, Whitaker-Menezes D, Castello-Cros R. (2010) The reverse Warburg effect glycolysis inhibitors prevent the tumor promoting effects of caveolin-1 deficient cancer associated fibroblasts. , Cell Cycle 9(10), 1960-1971.
- 87.Pavlides S, Tsirigos A, Vera I. (2010) Loss of stromal caveolin-1 leads to oxidative stress, mimics hypoxia and drives inflammation in the tumor microenvironment, conferring the "reverse Warburg effect": a transcriptional informatics analysis with validation. , Cell Cycle 9(11), 2201-2219.
- 88.Guido C, Whitaker-Menezes D, Capparelli C. (2012) Metabolic reprogramming of cancer-associated fibroblasts by TGF-beta drives tumor growth connecting TGF-beta signaling with "Warburg-like" cancer metabolism and L-lactate production. , Cell Cycle 11(16), 3019-3035.
- 89.Sonveaux P, Vegran F, Schroeder T. (2008) Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. , Journal of Clinical Investigation 118(12), 3930-3942.
- 90.Vegran F, Boidot R, Michiels C, Sonveaux P, Feron O. (2011) Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-kappa B/IL-8 pathway that drives tumor angiogenesis. , Cancer Research 71(7), 2550-2560.
- 91.Ahearne M J, Willimott S, Pinon L. (2013) Enhancement of CD154/IL4 proliferation by the T follicular helper (Tfh) cytokine, IL21 and increased numbers of circulating cells resembling Tfh cells in chronic lymphocytic leukaemia. , British Journal of Haematology 162(3), 360-370.
- 92.Ghia P, Circosta P, Scielzo C. (2005) Differential effects on CLL cell survival exerted by different microenvironmental elements. Chronic Lymphocytic Leukemia. 294, 135-145.
- 93.Willimott S, Wagner S D. (2012) Stromal cells and CD40 ligand (CD154) alter the miRNome and induce miRNA clusters including, miR-125b/miR-99a/let-7c and miR-17-92 in chronic lymphocytic leukaemia. , Leukemia 26(5), 1113-1116.
- 94.Zhu H, Shyh-Chang N, Segre A V. (2011) . The Lin28/let-7 Axis Regulates Glucose Metabolism. Cell 147(1), 81-94.
- 95.Jiang S, Zhang L-F, Zhang H-W. (2012) A novel miR-155/miR-143 cascade controls glycolysis by regulating hexokinase 2 in breast cancer cells. , Embo Journal 31(8), 1985-1998.
- 96.Dormond O, Contreras A G, Meijer E. (2008) CD60-induced signaling in human endothelial cells results in mTORC2-and akt-dependent expression of vascular endothelial growth factor in vitro and in vivo. , Journal of Immunology 181(11), 8088-8095.
- 97.Zent C S, LaPlant B R, Johnston P B. (2010) The treatment of recurrent/refractory chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL) with everolimus results in clinical responses and mobilization of CLL cells into the circulation. , Cancer 116(9), 2201-2207.
- 98.Dufort F J, Bleiman B F, Gumina M R. (2007) Cutting edge: IL-4-mediated protection of primary B lymphocytes from apoptosis via stat6-dependent regulation of glycolytic metabolism. , Journal of Immunology 179(8), 4953-4957.
- 99.Chu K, Boley K M, Moraes R, Barsky S H, Robertson F M. (2013) The paradox of E-cadherin: role in response to hypoxia in the tumor microenvironment and regulation of energy metabolism. , Oncotarget 4(3), 446-462.
- 100.Lutzny G, Kocher T, Schmidt-Supprian M. (2013) Protein Kinase C-beta-dependent activation of NF-kappa B in stromal cells is indispensable for the survival of chronic lymphocytic leukemia B cells in vivo. , Cancer Cell 23(1), 77-92.
- 101.Zhang W, Trachootham D, Liu J. (2012) Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. , Nature Cell Biology 14(3), 276-286.
- 102.Fajas L. (2013) Re-thinking cell cycle regulators: the cross-talk with metabolism. Frontiers in oncology. 3, 4-4.
Cited by (1)
- 1.Mirabilii Simone, Ricciardi Maria Rosaria, Tafuri Agostino, 2020, mTOR Regulation of Metabolism in Hematologic Malignancies, Cells, 9(2), 404, 10.3390/cells9020404