
Abstract
Canavan disease (CD) is a globally occurring but rare human spongiform leukodystrophy that is associated with inborn errors affecting the activity of aspartoacylase (ASPA), an enzyme highly expressed in oligodendrocytes that hydrolyzes N-acetylaspartate (NAA). Lack of ASPA activity is associated with the inability of oligodendrocytes to build or maintain axon-enveloping myelin sheaths. The primary source of NAA in brain is neurons, cells that synthesize but cannot catabolize it. Neurons also synthesize N-acetylaspartylglutamate (NAAG) from NAA and glutamate but cannot catabolize this substance as well. For their metabolism, these substances are released to extracellular fluid and are metabolized by oligodendrocyte ASPA and astrocyte NAAG peptidase respectively. A hypothesis developed suggested that the cause of the leukodystrophy component in CD was due to release of NAAG by neurons at white matter nodes of Ranvier, its catabolism by astrocytes forming NAA and increased osmotic-hydrostatic pressure as a result of its buildup at these nodes due to the lack of ASPA activity. In this communication, we provide evidence supporting this hypothesis and comment on the cause and possible cure for human CD.
Author Contributions
Academic Editor: Bobbie-Jo M. Webb-Robertson, Senior Research Scientist Pacific Northwest National Laboratory Computational Biology and Bioinformatics Richland , WA , USA
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2017 Shitao Li, et al
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Introduction
Rescuing Canavan Disease
We bring attention to a recent study 1 in which a human adeno-associated virus genetic aspartoacylase (ASPA) construct was serendipitously inserted into the “wrong” cell and by redirecting its metabolic processing rescued a murine model of Canavan disease (CD). After achieving an overall cure including enhanced motor performance, these authors were prompted to “hypothesize” that ASPA expression in a non-oligodendrocyte glial cell, astrocytes, might be involved. This was subsequently ascertained using appropriate cell markers. In this communication, we provide metabolic, physiological and cellular contexts as well as a plausible mechanism for evaluating this remarkable finding.
The Nature of Canavan Disease
CD is an autosomal recessive disease due to inborn errors resulting from more than 100 different mutations in which oligodendrocyte expressed ASPA is inactive 1, 2, 3. CD is a rare disease in that there are only several hundred human cases worldwide at any given time. Spatially, it is distributed among all races of the world, but is especially prevalent in the Ashkenazi Jewish population of northern Russia. ASPA and N-acetyl-L-aspartate (NAA), its natural substrate synthesized by neurons and maintained at high mM levels in both gray matter (GM) and white matter (WM), are clearly important for normal brain function as evidenced by these inborn metabolic errors. NAA is present in every human brain thus far examined except one 4, and in almost every other vertebrate brain examined. CD is manifested clinically as a spongiform leukodystrophy and is characterized by early onset, megalocephaly and a progressive loss of functions, generally leading to early mortality 2. Children with CD may appear relatively normal at birth but fail to reach typical milestones in development. An early subjective sign is the loss of ability to maintain their head in an upright position when being held. Other important clinical characteristics of CD are the buildup of high mM concentrations of NAA in brain, signs of WM pathology and presence of high concentrations of NAA in urine. Many therapeutic approaches to treatment of CD have been tried but with little success. Among these are the use of lithium in both animals and humans. Lithium treatment rapidly brings brain and urine NAA down to normal levels, but fails to achieve rescue of the spongiform demyelination in CD 2. There are still rarer mild cases of CD, with some individuals surviving for decades and having normal or near normal functionality although presenting with elevated NAA in brain and urine. The values for residual enzyme activity were derived from transfecting HEK293 cells with mutated genes and measuring residual expressed ASPA activity against the normal gene 3. These cases, although imperfectly associated with the amount of residual activity of the ASPA enzyme protein 3, are highly significant in that they show (1) that an elevated level of NAA in whole brain by itself does not cause CD, and (2) that a small amount of residual ASPA activity (<1 to 12.4 %) may rescue CD. The imperfect association between residual ASPA protein activity and mild cases of human CD may be explained by the presence of a second non-specific acylase that has been observed to be expressed by cultured rat astrocytes and that has some activity against NAA 5.
The Tri-Cellular Metabolism of NAA
ASPA and NAA are part of a unique tri-cellular metabolic system in brain wherein NAA is made in neurons from acetyl Co-enzyme A and L-aspartate by NAA synthase and is then used to synthesize an L-glutamate (Glu) adduct, the neurotransmitter N-acetylaspartylglutamate (NAAG), via NAAG synthase 6. Neurons cannot further metabolize either of these substances and for their hydrolysis they must be released to extracellular fluid (ECF). NAAG is targeted to the metabotropic glutamate receptor 3 (mGluR3) on the surface of astrocytes and upon docking is cleaved into Glu and NAA by NAAG peptidase. The Glu can be converted into glutamine for recycling to neurons, but the NAA product cannot be further hydrolyzed by astrocytes since they do not express ASPA 7 and it must be released to oligodendrocytes which do express this enzyme. This system requires the coordinated functioning of four enzymes, two anabolic and two catabolic, expressed in three different cell types and a specific receptor for its completion.
Inborn Errors in NAA and NAAG Metabolism and their Related Human Brain Pathologies
Both NAA and NAAG are dynamic and turn over every 14-16 h. While the function of this entire system is as yet unclear, one hypothesis suggests that the role of the astrocyte surface mGluR3-NAAG peptidase complex is to liberate Glu from NAAG to signal via astrocytes a neuron’s ongoing requirements for energy and oxygen in order to increase focal blood flow 8, 9. NAAG is a neurotransmitter specifically targeted to astrocytes, the key cellular components in neurovascular coupling in both GM and WM. If this is the case, then any reduction in this process could be detrimental, and any enhancement beneficial to neuron function. The complete absence of ASPA activity results in the severe form of human CD 3 and the complete absence of NAA synthase activity in a single known human case results in hypoacetylaspartia, an inborn error where neither NAA nor NAAG are produced 10. In both of these human conditions, there are profound clinically observed negative consequences. However, in rodents, the effects of similar enzyme deficits, natural or engineered, are much less severe.
Discussion
The Cause of CD; the Astrocyte Hypothesis
Since the buildup of NAA in whole brain did not appear to be the cause the CD syndrome, it was hypothesized that astrocytes in WM could be a source of NAA and the “missing link” in the process resulting in the spongiform leukodystrophy component in CD 11. This was based on two additional observations. First, NAA was not a neurotransmitter and there was no discernable reason for it to be released into limited WM ECF space, and second, that all the elements associated with NAAG synthesis, release, docking and catabolism were present in WM nodes of Ranvier. Based on these observations it was reasoned that the neurotransmitter NAAG liberated at nodes of Ranvier in WM would first dock with the astrocyte mGluR3 receptor, be catabolized by astrocyte NAAG peptidase forming Glu and NAA, the latter which would then be released within the node, catabolized by oligodendrocyte ASPA and recycled. However, in CD it was proposed that NAA formed from NAAG could not be hydrolyzed by juxtaposed myelinating oligodendrocytes. Consequently, NAA would build up in limited ECF space at these nodes, penetrating between extant oligodendrocyte myelin layers and producing an osmotic-hydrostatic pressure effect resulting in formation of destructive edematous spongiform vacuoles between myelin layers. Humans are born with some degree of myelination and once this destructive process is established in the first few months of life, re-myelination is highly unlikely. This hypothesis fits well with, and explains the known early onset, clinical pathology, progression and final outcome of CD. The proposed normal NAA-NAAG tri-cellular metabolic sequence at a node of Ranvier is shown in figure 1.
Figure 1.Cartoon showing hypothesized site of liberation of NAAG at neuron axonal nodes. NAA is generated by neuron NAA synthase and then NAAG via NAAG synthase. NAAG is found in highest concentration in WM axons. In GM, there is evidence that NAAG is released and docks with astrocyte mGluR3 and is then catabolized by astrocyte NAAG peptidase producing NAA and Glu. The Glu activates release of astrocyte prostaglandin messengers that signal the vascular system to increase blood flow, and the NAA is catabolized by oligodendrocyte ASPA forming Asp and Ac for recycling. This system is also present in WM and considered to function similarly in order to service axonal metabolic needs. Nodes are spaced approximately 1 mm (1000 μm) apart, and are about 2-3 μm wide (about 10 nodes/cm). In this illustration, internodes and compact myelin are highly compressed. NAAG, red filled circles; NAA, black filled dots. Adapted from Baslow and Guilfoyle (2009).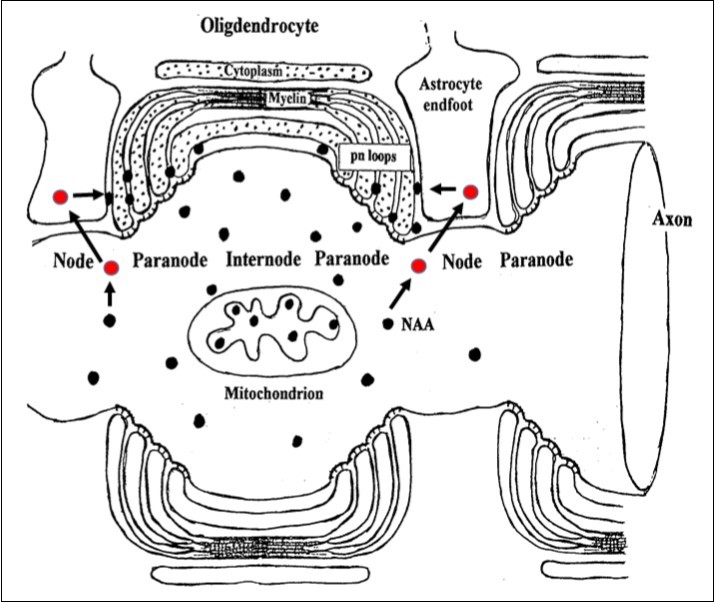
Genetic Engineering and CD
Previous genetic engineering attempts to treat human CD have been aimed at oligodendrocytes, the cells that naturally express ASPA, and neurons which do not. As reported 1, there has been a breakthrough in treatment of murine CD using a novel third generation vector construct gene therapy approach that inserts the normal gene into astrocytes rather than into oligodendrocytes. This appears to be of major importance in rescuing CD in that ASPA activity is introduced at the end of the NAAG metabolic chain within the same cell that hydrolyzes NAAG forming NAA after it has docked with and activated the mGluR3 astrocyte receptor, thus conserving its physiological function in neurovascular coupling. Astrocytes are also a prime candidate for gene insertion in that their endfeet surround about 90% of capillaries and they are among the first brain parenchymal cells to encounter a viral-gene blood-borne construct in both GM and WM.
A Long-Term Single Treatment Cure for CD?
The breakthrough treatment reported for murine CD is novel in that it is a one-time treatment that bypasses the role of oligodendrocytes by inserting the engineered ASPA gene directly into astrocytes. In GM it could be expected to eliminate the buildup of NAA in ECF from any source. In WM it would be expected to eliminate buildup of nodal NAA produced from NAAG by astrocytes thus resolving the problem of demyelination as a result of increased osmotic pressure due to formation of NAA-containing fluid-filled vacuoles between myelin sheaths. This advance in treatment does not prove, but strongly supports the hypothesis that the underlying cause of the CD syndrome is not the buildup of NAA in whole brain, but the edematous spongiform leukodystrophy resulting from NAA buildup in restricted WM space at nodes of Ranvier. Evidence supporting the astrocyte hypothesis of the cause of CD spongiform demyelination is as follows:
Mild cases of CD show that buildup of NAA in whole brain does not cause CD.
Mild cases of CD show that < 10 % of residual ASPA activity can rescue CD.
Astrocytes are integral components of WM nodes of Ranvier and the neurovascular coupling mechanism for removal of excess K+ and waste products as well as for supply of energy and metabolites to myelinated axons.
The complete NAA-NAAG tri-cellular metabolic system (neuron-astrocyte-oligodendrocyte) is present in close proximity at every node of Ranvier.
NAAG is a neurotransmitter specifically targeted to the astrocyte mGluR3 receptor.
NAAG is present at highest concentrations in neuron axons in WM.
NAAG is catabolized by astrocyte NAAG peptidase forming Glu and NAA.
The Glu product activates astrocyte neurovascular coupling via second messengers.
The NAA product is normally catabolized and recycled by oligodendrocyte ASPA.
In severe cases of CD, ASPA activity is very low and NAA builds to highest mM levels.
Redirecting a normal ASPA gene into astrocytes instead of adjacent oligodendrocytes normalizes NAA levels in whole brain and rescues a murine model of CD.
Conclusions
CD, its Etiology and A Possible Human Treatment
In this commentary, we have addressed the etiology of the CD syndrome and make the case that it is primarily an osmotic demyelinating disease caused by the buildup of NAA at nodes of Ranvier in WM. We have also reasoned that blocking the buildup of NAA in WM should resolve this problem. The report that redirecting ASPA activity to astrocytes in a murine model via a genetic engineering treatment on postnatal day 1 rescues CD reinforces this conclusion. Based on the difference in lifespan between species, the window of opportunity for intervention in humans may be a few months rather than days. It is hoped that this treatment can be developed for human CD in the near future. If successful, it would constitute not only an unusual genetic engineering success as a result of placing an engineered enzyme construct in the wrong cell but at the right time to treat CD, but importantly as a potential model opening the door for treating other metabolic diseases by redirecting their metabolic processing.
Abbreviations Used:
ASPA, aspartoacylase; CD, Canavan disease; ECF, extracellular fluid; Glu, glutamate; GM, gray matter; mGluR3, metabotropic glutamate receptor 3; NAA, N-acetylaspartate; NAAG, N-acetylaspartylglutamate; WM, white matter
References
- 1.D J Gessler, Li D, Xu H, Su Q, Sanmiguel J et al. (2017) Redirecting N-acetylaspartate metabolism in the central nervous system normalizes myelination and rescues Canavan disease. JCI Insight . doi:. 10-1172.
- 2.M H Baslow, D N Guilfoyle. (2013) Canavan disease, a rare early-onset human spongiform leukodystrophy: Insights into is genesis and possible clinical interventions. , Biochimie 95, 946-956.
- 3.Mendes M I, D E Smith, Pop A, Lennertz P, M R Ojeda et al. (2017) Clinically distinct phenotypes of Canavan disease correlate with residual aspartoacylase enzyme activity. , Hum. Mutat doi:, 1002-23181.
- 4.Martin E, Capone A, Schneider J, Hennig J, Thorsten T. (2001) Absence of N-acetylaspartate in the human brain: Impact on neurospectroscopy ?. , Ann. Neurol 49, 518-521.
- 5.M H Baslow, R F Suckow, M J Berg, Marks N, Saito M et al. (2001) differential expression of carnosine, homocarnosine and N-acetyl-L-histidine hydrolytic activities in cultured rat macroglial cells. , J. Mol. Neurosci 17, 351-359.
- 6.M H Baslow. (2000) Functions of N-acetyl-L-aspartate and N-acetyl-L-aspartylglutamate in the vertebrate brain. Role in glial cell-specific signaling. , J. Neurochem 75, 453-459.
- 7.M H Baslow, Suckow R, Sapirstein V, B L Hungund. (1999) Expression of aspartoacylase activity in cultured rat macroglial cells is limited to oligodendrocytes. , J. Mol. Neurosci 13, 47-53.
- 8.Baslow M H, Nowak K, Hungund B L, Guilfoyle D N.VV Dyakin.(2005) 2-PMPA, a NAAG peptidase inhibitor, attenuates the BOLD signal in brain of anesthetized mice: Evidence of a link between NAAG release and hyperemia. , J. Mol. Neurosci 26, 1-16.
- 9.M H Baslow, D N Guilfoyle. (2016) Evidence that N-acetylaspartylglutamate is the astrocyte-targeted neurovascular coupling agent that regulates slow tonic control of brain blood flow. , J. Glycomics and Metabolism https://www.openaccesspub.org 1(1), 1-12.